FROM: FASEB J. 1999 (Jul); 13 (10): 1145–1155 ~ FULL TEXT
Regina Brigelius–Flohé, and Maret G. Traber
German Institute of Human Nutrition,
Bergholz–Rehbrücke,
Germany Department of Nutrition and Food Management,
Linus Pauling Institute,
Oregon State University,
Corvallis, Oregon 97330, USA.
Flohe.www.dife.deAlthough vitamin E has been known as an essential nutrient for reproduction since 1922, we are far from understanding the mechanisms of its physiological functions. Vitamin E is the term for a group of tocopherols and tocotrienols, of which alpha–tocopherol has the highest biological activity. Due to the potent antioxidant properties of tocopherols, the impact of alpha–tocopherol in the prevention of chronic diseases believed to be associated with oxidative stress has often been studied, and beneficial effects have been demonstrated. Recent observations that the alpha–tocopherol transfer protein in the liver specifically sorts out RRR–alpha–tocopherol from all incoming tocopherols for incorporation into plasma lipoproteins, and that alpha–tocopherol has signaling functions in vascular smooth muscle cells that cannot be exerted by other forms of tocopherol with similar antioxidative properties, have raised interest in the roles of vitamin E beyond its antioxidative function. Also, gamma–tocopherol might have functions apart from being an antioxidant. It is a nucleophile able to trap electrophilic mutagens in lipophilic compartments and generates a metabolite that facilitates natriuresis. The metabolism of vitamin E is equally unclear. Excess alpha–tocopherol is converted into alpha–CEHC and excreted in the urine. Other tocopherols, like gamma– and delta–tocopherol, are almost quantitatively degraded and excreted in the urine as the corresponding CEHCs. All rac alpha–tocopherol compared to RRR–alpha–tocopherol is preferentially degraded to alpha–CEHC. Thus, there must be a specific, molecular role of RRR–alpha–tocopherol that is regulated by a system that sorts, distributes, and degrades the different forms of vitamin E, but has not yet been identified. In this article we try to summarize current knowledge on the function of vitamin E, with emphasis on its antioxidant vs. other properties, the preference of the organism for RRR–alpha–tocopherol, and its metabolism to CEHCs.
From the FULL TEXT Article:
BACKGROUND
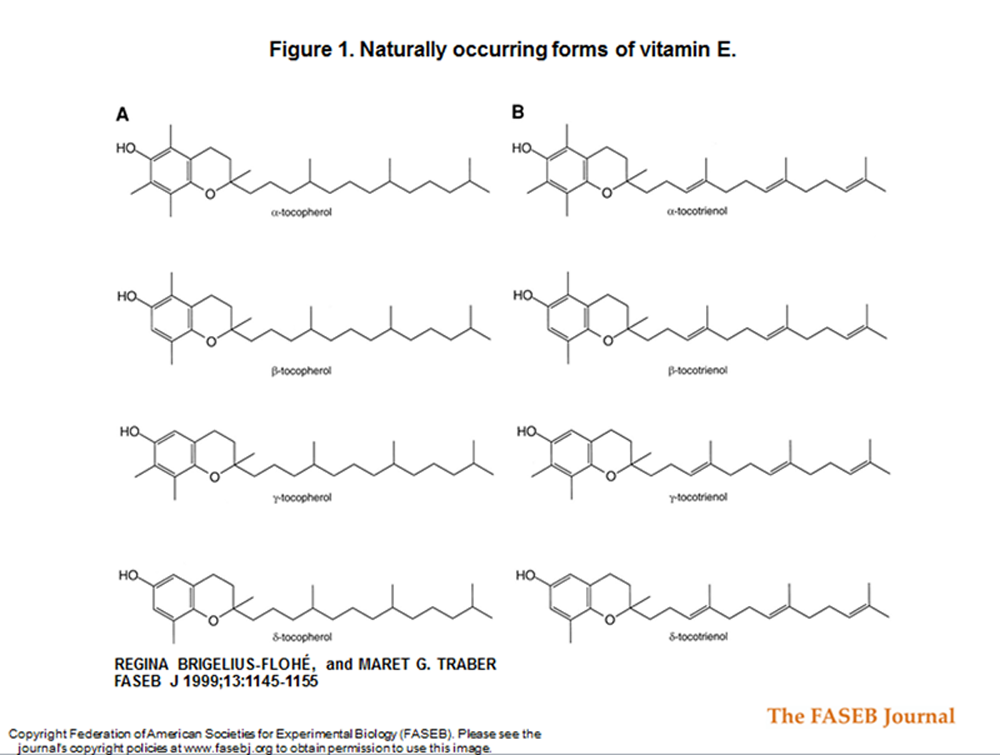
Figure 1 VITAMIN E IS a term that encompasses a group of potent, lipid–soluble, chain–breaking antioxidants. Structural analyses have revealed that molecules having vitamin E antioxidant activity include four tocopherols (α, β, γ, δ) and four tocotrienols (α, β, γ, δ); see Fig. 1 A, B. [1] One form, α–tocopherol, is the most abundant form in nature [2], has the highest biological activity based on fetal resorption assays [3–5], and reverses vitamin E deficiency symptoms in humans. [6–10] The molecular functions fulfilled specifically by α–tocopherol have yet to be fully described, but it is unlikely they are limited to general antioxidant functions.
In 1922, Evans and Bishop [11] discovered vitamin E—a micronutrient essential for reproduction in rats. It was rediscovered in the 1950s as factor 2 by Klaus Schwarz [12] and placed in the context of cellular antioxidant systems, together with sulfur amino acids (factor 1) and selenium (factor 3). Vitamin E subsequently proved to be effective in preventing lipid peroxidation and other radical–driven oxidative events. [13–15]
The antioxidant activity of vitamin E has persuaded many groups to study its ability to prevent chronic diseases, especially those believed to have an oxidative stress component such as cardiovascular diseases, atherosclerosis, and cancer. Epidemiological studies [16, 17] have reported that high vitamin E intakes are correlated with a reduced risk of cardiovascular diseases, whereas intakes of other dietary antioxidants (such as vitamin C and β–carotene) are not, suggesting that vitamin E plays specific roles beyond that of its antioxidant function.
The possibility that vitamin E has an ameliorative effect in chronic disease has spurred interest in determining its specific molecular functions and whether these are related to its antioxidant function. This paper seeks to describe the current knowledge of vitamin E function: What are its specific antioxidant functions, its role in cell signaling, its recognition by the α–tocopherol transfer protein, and its metabolism? These important functions are discussed in relationship to human vitamin E deficiency, normal metabolism, and chronic disease.
FUNCTIONS
Antioxidant function Vitamin E functions as a chain–breaking antioxidant that prevents the propagation of free radical reactions. [13, 14, 18–21]
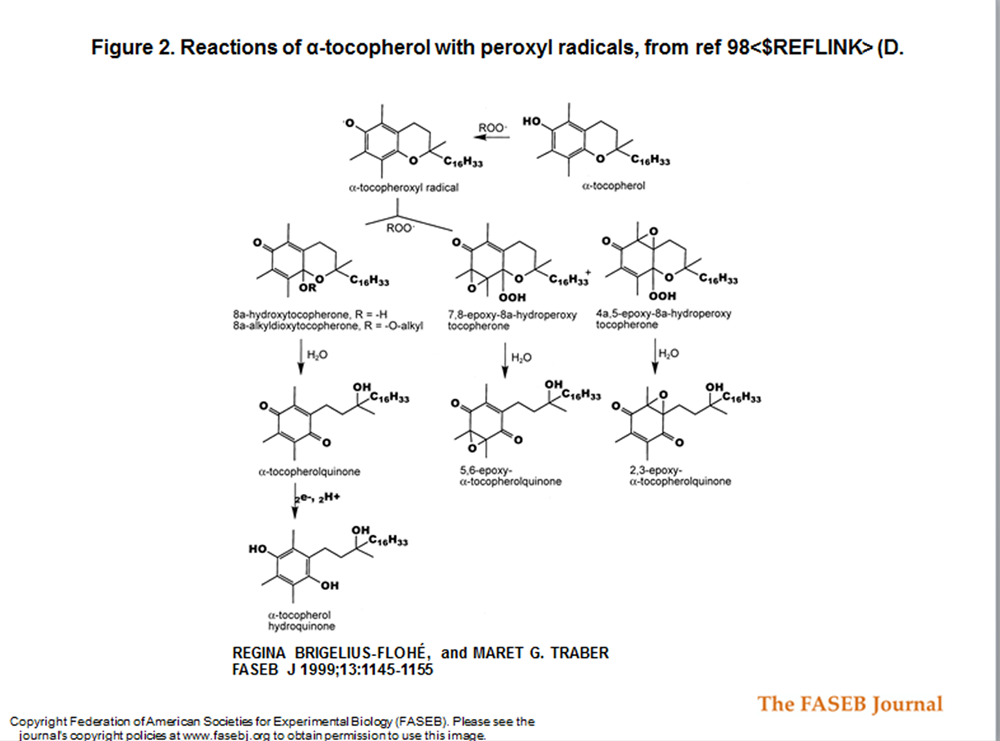
Figure 2 To induce lipid peroxidation, Ham and Liebler [22] perfused male Sprague–Dawley rat livers with 2 mM tert–butylhydroperoxide (t–BuOOH)2 for 10 min. t–BuOOH induced oxidation of α–tocopherol to α–tocopherol quinone, α–tocopherol hydroquinone, 2,3–epoxy–α–tocopherol quinone, and 5,6–epoxy–α–tocopherol quinone (Figure 2). Isolated mitochondria from these livers exhibited increased state 3 and state 4 respiration and a decline in the respiratory control ratio. However, in livers from rats given supplementary vitamin E (which contained 7– to 10–fold higher vitamin E levels than controls), lipid peroxidation and metabolic changes induced by t–BuOOH were decreased; t–BuOOH–induced increase in state 4 respiration did not occur and the respiratory control ratio was maintained. The relative extent of α–tocopherol oxidation, oxidation products formed, and distribution in whole liver and isolated mitochondria was similar in the vitamin E–supplemented and unsupplemented animals. These data suggest that the ‘extra’ vitamin E prevented mitochondrial dysfunction in the face of severe oxidative stress.
In vivo antioxidant activity In 1996, the Cambridge Heart Antioxidant Study (CHAOS) [23] reported in over 2000 patients with angiographically proven coronary atherosclerosis that vitamin E supplementation (400–800 IU/day) for slightly under 2 years significantly (P<0.005) reduced the incidence of cardiovascular death and nonfatal myocardial infarction by 77%. Decreases in lipid peroxidation of low density lipoproteins (LDL) have been assumed to be the mechanisms for this result. However, tissue responses to oxidative stress may be important. In a Japanese trial of 60 patients with coronary spastic angina, treatment for 30 days with a daily dose of 300 mg α–tocopherol resulted in a significant improvement (P<0.001) of impaired endothelium–dependent vasodilation concomitant with a reduction of plasma TBARS (thiobarbituric acid–reactive substrate), a marker of lipid peroxidation. [24]
Demonstration of free radical damage and its prevention by vitamin E in vivo have lagged because of a lack of sensitive analytical techniques. This, however, has recently changed; quantification of F2–isoprostanes, isomers of prostaglandin F2, has been suggested by a number of investigators as a reliable index of in vivo free radical generation and oxidative lipid damage. F2–isoprostanes are formed in membranes from arachidonyl–containing lipids by cyclooxygenase enzymes, as well as during free radical–catalyzed lipid peroxidation. [25–27] In studies using experimental animals, F2–isoprostanes increased in plasma and tissues as a result of vitamin E deficiency. [28] Furthermore, in an animal atherosclerosis model (the apoE–deficient mouse), vitamin E supplementation not only suppressed F2–isoprostane production but also decreased atherosclerotic lesion formation. [29]
8–epi–prostaglandin F2α (8–iso–PGF2α) is one of the more abundant F2–isoprostanes produced in vivo in humans. [30] 8–iso–PGF2α excretion is depressed in humans by antioxidant vitamins [31, 32], but not by the nonspecific cyclooxygenase inhibitor, aspirin. [32] In hypercholesterolemic subjects, 8–iso–PGF2α and 11–dehydro–thromboxane B2 (a marker of platelet activation) were elevated, but urinary excretion was unaffected by low–dose aspirin and indobufen, a reversible cyclo–oxygenase inhibitor, whereas these agents decreased thromboxane biosynthesis. [33] In contrast, vitamin E supplementation resulted in a dose–dependent reduction in 8–iso–PGF2α excretion and decreased sensitivity of LDL to in vitro oxidation [33]. Davi et al. [33] suggest that pharmacological vitamin E doses in humans may decrease an aspirin–insensitive prothrombotic factor involved in myocardial infarction. This hypothesis provides an alternative mechanism for the beneficial effects of vitamin E seen in the CHAOS study. [23]
Pro–oxidant activity The antiatherogenic results conflict with pro–oxidative vitamin E effects observed in vitro. It has to be considered that vitamin E, like every redox–active compound, may exert anti– and pro–oxidative effects depending on the reaction partners present. Pro–oxidative functions of α–tocopherol have been demonstrated in LDL isolated from healthy volunteers [34] and a patient with a defect in the α–TTP gene. [35] The importance of a pro–oxidant role of vitamin E in vivo has yet to be demonstrated. Certainly in the presence of other coantioxidants, including ascorbic acid and ubiquinol, vitamin E does not have a prooxidant function. This topic is more fully discussed in the review by Upston et al. [36]
Specific chemical role for γ–tocopherol? A role distinct from oxygen radical scavenging has been proposed for γ–tocopherol. In contrast to α–tocopherol, γ–tocopherol is a powerful nucleophile that traps electrophilic mutagens in lipophilic compartments. [37–39] It thus complements glutathione, which similarly scavenges electrophilic mutagens in the aqueous phase of the cell. An electrophilic mutagen prone to react with γ–tocopherol is peroxynitrite. Thus, γ–tocopherol may protect lipids, DNA, and proteins from peroxynitrite–dependent damage.
Hoglen et al. [40] have shown that the reaction of peroxynitrite with γ–tocopherol in vitro results in the formation of four major products: 2,7,8–trimethyl–2–(4,8,12–trimethyldecyl)–5–nitro–6–chromanol (NGT), 2,7,8–trimethyl–2–(4,8,12–trimethyldecyl)–5,6–chromaquinone (tocored), and two diastereomers of 8 α–(hydroxy)–γ–tocopherone. However, when γ–tocopherol was reacted with the nitrating agent NO2+ BF4–, the major product detected was γ–tocopheryl quinone, a product that was not detected in reactions involving peroxynitrite. Since the product distribution after oxidation with NO2+ BF4– differed substantially from that after oxidation with peroxynitrite, NO2+ appears not to be the principal species involved in NGT formation. Nitration of γ–tocopherol may involve either peroxynitrite or some peroxynitrite–derived oxidant other than NO2–. Because of its stability and formation as a novel product of the reaction between γ–tocopherol with peroxynitrite, Hoglen et al. [40] suggest that NGT may be a useful in vivo marker for peroxynitrite interactions with γ–tocopherol.
Vitamin E deficiency, antioxidant function, and neurological dysfunction In humans, severe vitamin E deficiency leads to neuromuscular abnormalities characterized by spinocerebellar ataxia [8, 41–44] and myopathies. [9, 45] The peripheral neuropathy likely occurs due to free radical damage to the nerves and a dying back of the sensory neurons. [46] Similarly, vitamin E deficiency anemia occurs, largely in premature infants, as a result of free radical damage. [47] Diminished erythrocyte life span [48, 49] and increased susceptibility to peroxide–induced hemolysis are apparent not only in severe deficiency, but also in marginal vitamin E deficiency in hypercholesterolemic subjects. [50]
Overt vitamin E deficiency occurs only rarely in humans and virtually never as a result of dietary deficiencies. Vitamin E deficiency does occur as a result of genetic abnormalities in the α–tocopherol transfer protein (α–TTP) and as a result of various fat malabsorption syndromes, as reviewed in ref 51. The frequency of human vitamin E deficiency as a result of α–TTP defects is unknown.
Patients with familial isolated vitamin E deficiency, an inborn genetic defect in the gene for the α–tocopherol transfer protein [52, 53], have dramatically reduced plasma vitamin E levels and neurological disorders characteristic of vitamin E deficiency such as cerebellar ataxia, dysarthria, absence of deep tendon reflexes, vibratory and proprioceptive sensory loss, and positive Babinski sign. [52]
The deficiency symptoms associated with this syndrome can be ameliorated when these patients are given doses of vitamin E of up to 2000 mg per day. [8–10] Also, symptoms of vitamin E deficiency caused by chronic liver disease, fat malabsorption, or abetalipoproteinemia are ameliorated by high doses of vitamin E. [7, 54] None of the curative dosages can be achieved by an optimized dietary regimen; the patients must consume vitamin E supplements.
NONANTIOXIDANT FUNCTIONS?
Cellular signaling The role of vitamin E in cellular signaling, especially in relation to protein kinase C, has been studied intensively by Azzi’s group. [55] α–Tocopherol inhibits smooth muscle cell proliferation [56], decreases protein kinase C activity [57], increases phosphoprotein phosphatase 2A activity [58], and controls expression of the α–tropomyosin gene. [55] These functions are not related to vitamin E antioxidant action because β–tocopherol, which has a similar antioxidant activity, does not perform any of these functions; it actually abrogates the α–tocopherol effect.
α–Tocopherol effects on protein kinase C inhibition have also been reported in human platelets [59], diabetic rat kidney [60, 61], and human monocytes. [62] The mechanism of protein kinase C inhibition by α–tocopherol may be attributable in part to its attenuation of the generation of membrane–derived diacylglycerol, a lipid that activates protein kinase C translocation and activity. [63, 64] Cachia et al. [65] have also suggested that the inhibition of protein kinase C activity is not due directly to the antioxidant capacity of α–tocopherol, but requires the integration of α–tocopherol into a membrane structure, and is likely due to the direct interaction between α–tocopherol and protein kinase C in the cell membrane.
α–Tocopherol modulates the in vitro expression of some significant proteins/enzymes in various cell types involved in atherogenesis. [66] Vitamin E enrichment of endothelial cells down–regulates the expression of intercellular cell adhesion protein and vascular cell adhesion molecule–1, thereby reducing the oxidized LDL–induced adhesion of white cells to the endothelium. [67]
Recent advances in the area of the arachidonic acid cascade have also demonstrated that α–tocopherol can regulate these pathways, and its effect is not always shared by other vitamin E forms; see review. [66] Vitamin E up–regulates the activities of cytosolic phospholipase A2 [68, 69] and cyclooxygenase. [69] The enhanced activity of these two rate–limiting enzymes in the arachidonic acid cascade provides a mechanism for the observation that vitamin E dose–dependently enhances release of prostacyclin, a potent vasodilator and inhibitor of platelet aggregation. [70–74]
Infertility Vitamin E prevents loss of spermatogenesis in males and the failure to retain zygotes in female rats. [11] Male infertility also results from selenium deficiency, and could thus be envisaged to support a general antioxidant function of vitamin E in the reproductive system. A synergistic effect of vitamin E and selenium in the protection of biomembranes from oxidative attack has been widely discussed. Vitamin E is known to readily reduce alkyl peroxy radicals of unsaturated lipids [75], thereby generating hydroperoxides that are reduced by the selenoperoxidases, in particular by phospholipid hydroperoxide glutathione peroxidase. [76, 77] Correspondingly, vitamin E and selenium can substitute for each other or at least act synergistically in pathogenic phenomena arising from oxidative stress. [78] Surprisingly, however, vitamin E was not able to replace selenium in the prevention of functional and structural alterations of spermatozoa. [79] This observation suggests distinct roles for both micronutrients. The specific role of selenium in spermatogenesis appears to be related to phospholipid hydroperoxide glutathione peroxidase, which is expressed depending on the developmental state of spermatids [80] and seems to be converted into a structural component in the midpiece of mature spermatozoa. [81] The molecular role of α–tocopherol in this context remains undefined.
METABOLISM
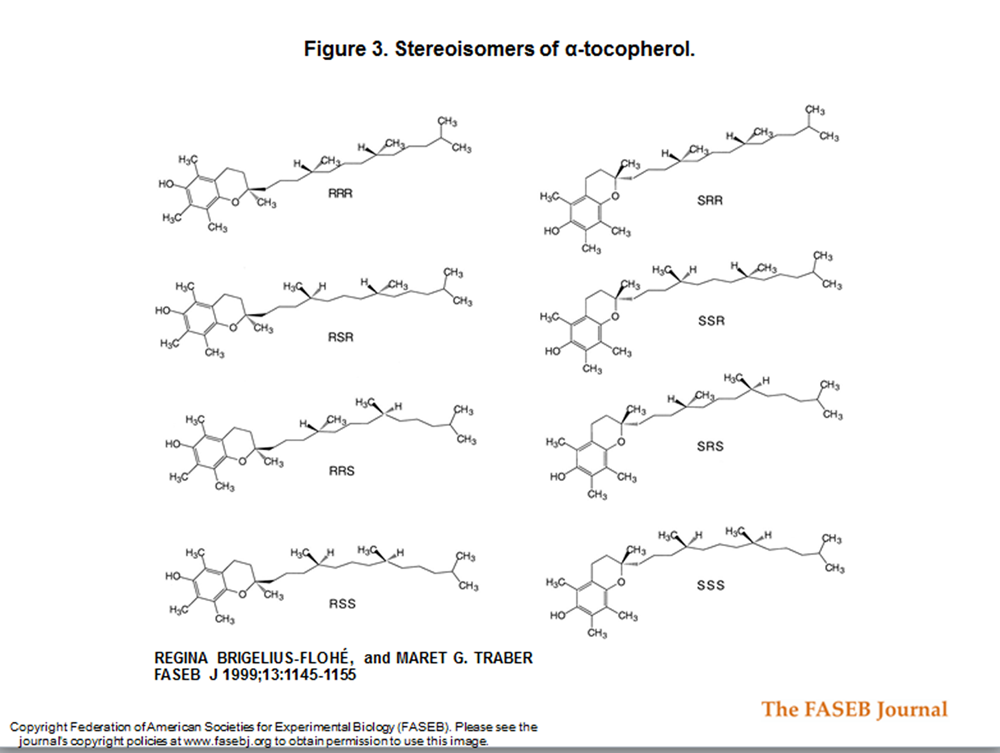
Figure 3 Biopotency and bioavailability Commercially available vitamin E supplements usually contain only α–tocopherol, provided either unesterified or usually as the ester of acetate, succinate, or nicotinate. Supplements can contain either the natural RRR– or the synthetic (all rac) α–tocopherol. all rac α–tocopherol consists of all eight possible stereoisomers arising from the three chiral centers, the C2 in the chroman ring and C4' and C8' in the phytyl tail [82] (Figure 3). None of forms are bioequivalent; the stereoisomers have been tested in the rat resorption–gestation test and showed highly different biological activities. [83] Taking the activity of the RRR–α–tocopheryl acetate as 100%, the other forms had the following activities: RRS 90%, RSS 73%, SSS 60%, RSR 57%, SRS 37%, SRR 31%, and SSR 21%. [83] Nonetheless, analysis of tissue stereoisomers demonstrated that the 2R forms were predominant. [5, 84]
In humans given an equimolar mixture of free and esterified α–tocopherol labeled with different amounts of deuterium, the concentrations of α–tocopherol derived from the both forms were equal in plasma and red blood cells. [85] These results show that in humans, free and esterified α–tocopherol have the same bioavailability.
Determinants of biological activity The biological activity of natural RRR–α–tocopherol is higher than that of the synthetic all rac α–tocopherol and other natural forms of vitamin E. Differences in absorption, however, do not satisfactorily explain the differences in the specific activities of the isomers. Instead, the following possibilities and facts may account for their differential therapeutic profiles:1) distinct biological activity of the isomers themselves or their metabolites
2) different rates and/or modes of metabolism
3) compartmentalization by transport mechanisms specific for RRR–α–tocopherol, and
4) specific interaction of individual isomers with particular receptors.Vitamin E is absorbed in the intestine and enters the circulation via the lymphatic system. It is absorbed together with lipids, packed into chylomicrons, and transported to the liver with the chylomicrons and the remnants derived thereof (reviewed in [51]). This process is similar for all forms of vitamin E tested. Only after passage through the liver does α–tocopherol preferentially appear in the plasma. [51] Most of the ingested β–, γ–, and δ–tocopherol is secreted into bile or not taken up and excreted in the feces. [86]
The reason for the plasma preference for α–tocopherol is its specific selection by the hepatic α–tocopherol transfer protein (α–TTP). [87] α–TTP not only specifically sorts out the α form of all tocopherols but also has a preference for 2R–stereoisomers. Supplementation studies with differentially deuterated α–tocopherols revealed that the 2R epimers compared with the 2S epimers are preferentially retained in all tissues except the liver. [88, 89] Moreover, use of chiral HPLC techniques has demonstrated that plasma and tissues after supplementation with all rac α–tocopherol contain the 2R epimers. [5, 84, 90]
Plasma RRR–α–tocopherol incorporation is a saturable process. Plasma levels on vitamin E supplementation cease to increase at ~80 µM despite increasing dosages of up to 800 mg RRR– [91, 92] or 1320 mg all rac α–tocopherol [93] per day. A dose response study using deuterated RRR–α–tocopherol demonstrated that the limitation in plasma α–tocopherol concentration appears to be a result of the rapid replacement of circulating with newly absorbed α–tocopherol. [94] These data are consistent with kinetic analyses demonstrating that the entire plasma pool of α–tocopherol is replaced daily. [95]
Degradation
Products with chroman ring oxidation Due to the great interest in the antioxidant function of vitamin E, studies of metabolism have concentrated on metabolites resulting from oxidation of the chroman moiety. The main hepatic oxidation product was described as α–tocopheryl quinone derived from the reaction of the tocopheroxyl radical with a peroxyl radical (Fig. 2). [96] α–Tocopheryl quinone can be reduced to α–tocopheryl hydroquinone by NAD(P)H–dependent microsomal and mitochondrial enzymes. [97] The quinone and the hydroquinone have both been found in biological membranes treated with azo–bis(amidinopropane, a peroxyl radical generator. [98]
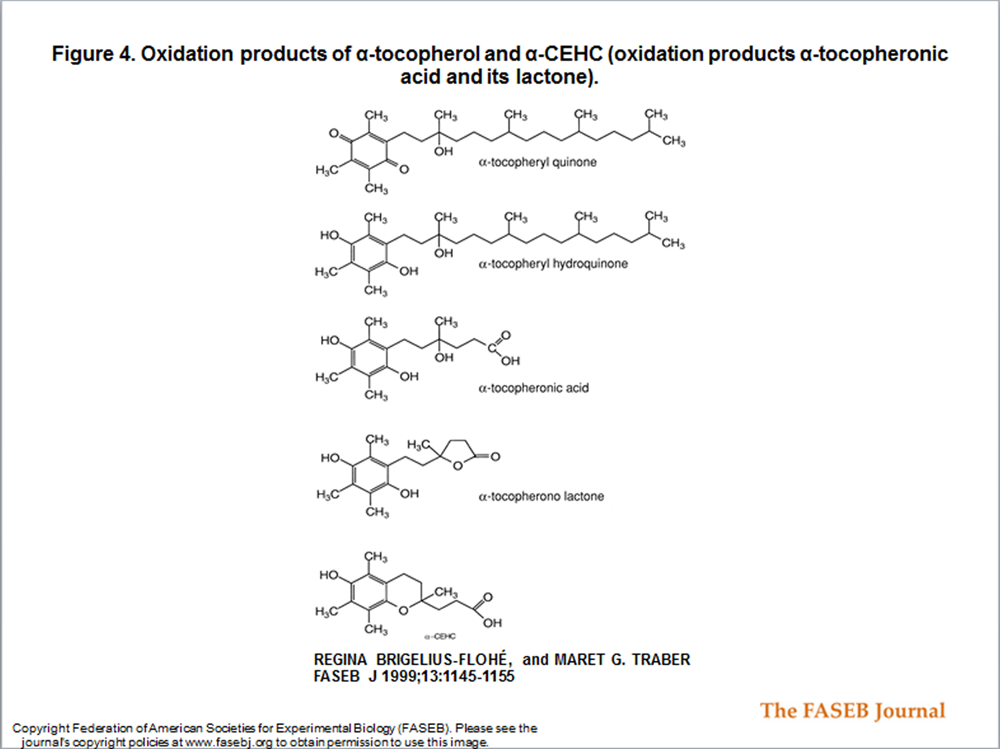
Figure 4 For decades, the only known urinary metabolites of α–tocopherol were the so–called Simon metabolites, first described in 1956. [99, 100] These metabolites, α–tocopheronic acid and its lactone, were found in increased amounts in the urine of subjects who consumed 3–5 g all rac α–tocopherol (Figure 4).
The chroman ring is opened in the Simon metabolites. Ring opening starts with the formation of the α–tocopheroxyl radical [96], i.e., when α–tocopherol has exerted its antioxidant function. The Simon metabolites were therefore taken as indicators that vitamin E had reacted as an antioxidant. It was paradoxical, however, why a high intake of vitamin E increased its oxidative destruction.
Metabolites with an intact chroman structure
α–CEHC Searching for urinary metabolites of vitamin E, another metabolite of α–tocopherol, 2,5,7,8–tetramethyl–2(2'–carboxyethyl)–6–hydroxychroman (α–CEHC), was identified [914, 101] (Fig. 4) . α–CEHC excretion was found to increase when a certain plasma level of RRR–α–tocopherol was exceeded. [91] The intact chroman structure of this metabolite indicates that α–CEHC is derived from α–tocopherol that has not reacted as an antioxidant. Therefore, it was concluded that the excretion of α–CEHC could be taken as an indicator of an adequate or excess α–tocopherol supply. [91]
To test why α–CEHC was not detected in the experiments of Simon et al. [100], the sample processing procedure was analyzed. When care was taken to avoid oxygenation during sample preparation, α–CEHC was detected in the sample [91, 101]; in the absence of argon or nitrogen protection, tocopheronolactone (e.g., Simon metabolite) appeared. In addition, when oxygen was bubbled through an α–CEHC solution, it was converted into tocopheronolactone within 8 h. [91] Therefore, metabolites previously identified in the urine after a high intake of α–tocopherol have probably derived during sample processing from α–CEHC. This appears to be a more reasonable explanation of why a higher excretion of the Simon metabolites is observed after a high intake of α–tocopherol than the explanation of an increased antioxidant function.
α–Tocopherol contains three chiral centers, one at the connection of the phytyl tail and the chroman ring (C2) and two in the phytyl tail itself. Because α–CEHC results from a tail shortening, it was of interest to investigate the conversion of all rac α–tocopherol compared with the conversion of RRR–α–tocopherol to the vitamin E metabolite. Therefore, six volunteers consuming equimolar doses of differentially deuterated RRR– and all rac α–tocopheryl acetate were studied. [102] After dosing, the plasma concentrations of deuterated RRR–α–tocopherol were twice those of deuterated all rac α–tocopherol, whereas urinary excretion of α–CEHC derived from the all rac form exceeded that of α–CEHC derived from the RRR form by a factor of 3 or 4. [102]
The plasma factor of 2 in favor of the RRR form has been observed in humans several times [89, 90, 102–105] and far exceeds the accepted ratio of biological activity of 1.36. This observation can only be explained by a preferential incorporation of those forms of α–tocopherol having the R–configuration at C2, which is 100% in the RRR form and 50% in the all rac form, respectively.
γ–CEHC The American diet contains large amounts of γ–tocopherol compared with the European diet due to the high consumption of soybean and corn oils by Americans. Despite the high intake of γ–tocopherol, plasma α–tocopherol is ~10–fold higher than γ–tocopherol, again demonstrating the preference of the sorting mechanism for α–tocopherol. Moreover, plasma γ–tocopherol has been shown to be replaced when the intake of α–tocopherol is increased. [106, 107]
Urinary γ–tocopherol excretion had not been investigated until the detection of a γ–tocopherol metabolite with an intact chroman structure and a shortened side chain. This metabolite was named LLU–α by Wechter et al. [108] for Loma Linda University metabolite–α, but it corresponds exactly to the α–tocopherol–derived α–CEHC [91] and therefore is referred to as γ–CEHC in the following discussion.
γ–CEHC was first detected as an endogenous natriuretic factor [108] in uremic patients, but is also found in normal human urine (109). The stereochemistry has been unequivocally established as S(+) by X–ray crystallography [108, 110], meaning that it was derived from 2R–γ–tocopherol without any epimerization at C–2. This finding is consistent with the metabolites formed from α– and δ–tocopherols. [91, 111, 112] The lack of an opening of the chroman ring can be taken as further evidence that oxidation of tocopherols is not a necessary step for urinary excretion of tocopherol metabolites.
Despite the usual higher α– to γ–tocopherol ratio in human plasma, urinary γ–CEHC excretion was higher than that of α–CEHC. [102] It has even been suggested that all of the ingested γ–tocopherol is converted to γ–CEHC [109], as has been shown for δ–tocopherol (see below). This is in contrast to α–CEHC excretion, which represents only a small part of the ingested α–tocopherol and shows that alternative routes of excretion may exist for α–tocopherol if it is not reincorporated into plasma. α–CEHC may represent the part of α–tocopherol that cannot be incorporated into VLDL either because of saturation or due to a stereochemistry that cannot be selected by α–TTP.
δ–CEHC δ–CEHC was the first urinary vitamin E metabolite to be detected that did not result from oxidative destruction of the chroman ring. [111] δ–CEHC was found in the urine of rats given tritium–labeled δ–tocopherol intravenously. About 50% of the given dose appeared as δ–CEHC in the urine. This means that a substantial portion of δ–tocopherol is degraded into δ–CEHC.
As originally suggested for δ–CEHC [111], CEHCs are probably formed according to the following degradation pathway: shortening of the side chain to three carbons by ω–oxidation and subsequent β–oxidation. [91, 101] The chroman ring is maintained during the metabolic process. The enzyme systems that degrade the side chains have yet to be identified. The reason for a suggested higher degradation of δ–tocopherol compared with α–tocopherol was explained by steric hindrance of the methyl group. But in view of today’s knowledge concerning the function of α–TTP, it is likely that α–tocopherol is salvaged and its metabolism and excretion are prevented by the function of α–TTP, while preferential degradation of unselected forms occurs. This hypothesis is supported by the observation that synthetic all rac α–tocopherol is more readily converted to α–CEHC than is natural RRR–α–tocopherol.
SUMMARY AND OUTLOOK
Three quarters of a century after its discovery, we are just beginning to understand vitamin E physiology. Identification of the various types of tocopherols and the availability of synthetic isomers were instrumental in defining particular effects, suggesting specific functions of the individual types and isomers. α–Tocopherol, the prominent component of the vitamin E complex, is unique in many respects. A sorting process exists only for the natural RRR–α–tocopherol; α–TTP–preferentially incorporates only the natural RRR–α–tocopherol into plasma. γ– and δ–tocopherols can practically substitute for α–tocopherol in in vitro antioxidant action and partially in the resorption–gestation assay, the differences being primarily due to the α–tocopherol–specific sorting system. An analogous in vivo lack of equivalence exists between RRR–α–tocopherol and all rac α–tocopherol. The sorting process is highly specific and does not tolerate the alteration in stereochemistry at C2. This preference prevents α–tocopherol from rapid ω–oxidation and thus keeps the excretion of the α–CEHC degradation product low. Only excess α–tocopherol seems to be converted to α–CEHC, whereas ingested γ– and δ–tocopherol may be almost quantitatively converted to their CEHCs and excreted in the urine. These findings suggest that there is a specific, molecular role for RRR–α–tocopherol.
α–Tocopherol also appears unique in regulating phosphorylation cascades. Such a role may be important in heart disease where cell adhesion, proliferation, and oxidant production may all be modified through vitamin E–sensitive pathways.
γ–Tocopherol is unique in being a potent nucleophile and generating a metabolite, γ–CEHC, with an intriguing pharmacological function, natriuresis. The former may contribute to the scavenging of electrophilic mutagens, the latter to the prevention of cardiovascular disease by lowering blood pressure.
In view of the significant differences in metabolism and, as a result, biopotency of the individual tocopherols, it is not surprising that many epidemiological and intervention studies aiming to elucide presumed vitamin E effects that did not take into account the metabolic differences between tocopherols remained inconclusive. Definite recommendation on the uses and dosages of tocopherols can only be expected from prospective intervention studies of individual tocopherols with defined isomers and stereochemistry.
Abbreviations:
α–CEHC, 2,5,7,8–tetramethyl–2(2'–carboxyethyl)–6–hydroxychroman
CHAOS, Cambridge Heart Antioxidant Study
8–iso–PGF2α, 8–epi–prostaglandin F2α
NGT, 2,7,8–trimethyl–2–(4,8,12–trimethyldecyl)–5–nitro–6–chromanol
t–BuOOH, tert–butylhydroperoxide
α–TTP, α–tocopherol transfer protein.

Return to the VITAMIN E Page
Since 4–21–2016