

Laboratory Science:
Human Parainfluenza Virus InfectionHuman Parainfluenza Virus Infection of the Airway Epithelium:
Viral Hemagglutinin-Neuraminidase Regulates Fusion
Protein Activation and Modulates Infectivity
This section is compiled by Frank M. Painter, D.C.
Send all comments or additions to: Frankp@chiro.org


FROM: Journal of Virology 2009 (Jul); 83 (13): 6900–6908 ~ FULL TEXT
Laura M. Palermo, Matteo Porotto, Christine C. Yokoyama, Samantha G. Palmer,
Bruce A. Mungall, Olga Greengard, Stefan Niewiesk, and Anne Moscona
Department of Pediatrics and of Microbiology and Immunology,
Weill Medical College of Cornell University,
515 East 71st Street, 6th Floor,
New York, NY 10021, USA.
Why was this study done?
This study was designed to help understand the progression of parainfluenza infection for the development of potential anti-viral strategies.
What This Study Found
The study suggested that the most direct strategy for preventing infection may be by interfering with the initial receptor binding of the virus to the host cell.
Three discrete activities of the paramyxovirus hemagglutinin-neuraminidase (HN) protein, receptor binding, receptor cleaving (neuraminidase), and triggering of the fusion protein, each affect the promotion of viral fusion and entry. For human parainfluenza virus type 3 (HPIV3), the effects of specific mutations that alter these functions of the receptor-binding protein have been well characterized using cultured monolayer cells, which have identified steps that are potentially relevant to pathogenesis. In the present study, proposed mechanisms that are relevant to pathogenesis were tested in natural host cell cultures, a model of the human airway epithelium (HAE) in which primary HAE cells are cultured at an air-liquid interface and retain functional properties. Infection of HAE cells with wild-type HPIV3 and variant viruses closely reflects that seen in an animal model, the cotton rat, suggesting that HAE cells provide an ideal system for assessing the interplay of host cell and viral factors in pathogenesis and for screening for inhibitory molecules that would be effective in vivo. Both HN's receptor avidity and the function and timing of F activation by HN require a critical balance for the establishment of ongoing infection in the HAE, and these HN functions independently modulate the production of active virions. Alterations in HN's F-triggering function lead to the release of noninfectious viral particles and a failure of the virus to spread. The finding that the dysregulation of F triggering prohibits successful infection in HAE cells suggests that antiviral strategies targeted to HN's F-triggering activity may have promise in vivo.
From the FULL TEXT Article:
Background
Paramyxoviruses are enveloped viruses that enter cells by fusing directly with the cell membrane. During entry, the viral surface glycoproteins hemagglutinin-neuraminidase (HN) (the receptor-binding molecule) and F (the fusion protein) cooperate in a highly specific way to mediate fusion upon receptor binding. To understand these mechanisms, elucidate how paramyxoviruses enter cells, and develop strategies to prevent or treat infection, we study human parainfluenza virus (HPIV), an important cause of croup and bronchiolitis in children. Our results have uncovered fundamental roles of the receptor-binding protein in paramyxovirus fusion and principles of coordinated interaction between the glycoproteins during the viral life cycle.
To understand how the diverse functions of the viral glycoproteins are regulated during the viral life cycle, we have used viruses bearing variant HN molecules with mutations at the binding/F-triggering site (and/or the primary receptor-binding site) to study how this molecule works to trigger F. [2, 3, 7, 10, 15, 18, 20] The correct timing of F activation (triggering) by HN is essential for entry. For infection to occur, triggering must occur only when F is in proximity to the target cell membrane, and we propose that the regulation of F triggering is essential for the survival of the virus. The outcome of infection is determined by the target cell's properties and its receptors, and specific mechanisms that are relevant to pathogenesis need to be tested using tissues that reflect the natural host. We therefore tested the hypothesis that a dysregulation of F triggering precludes successful infection in both a cotton rat model and the natural host airway epithelium.
For the cotton rat model, previous studies suggested that altered pathogenesis in HPIV infection might be caused by specific HN mutations. [24] The present detailed studies of the cotton rat using HN viral variants suggest that the extent of lung infection correlates with the ability of each variant to grow in vivo. The most striking finding is that the ability of the HN variants to grow in vivo is inversely related to their ability to fuse a monolayer of cultured cells. In order to understand the determinants of infection in the natural host, we therefore turned to a model that closely reflects the natural human host tissue, the human airway epithelium (HAE). This model utilizes a recently developed method for culturing primary HAE cells at an air-liquid interface, generating a differentiated, pseudostratified, mucociliary epithelium that faithfully represents the HAE. [16] The HAE model was previously used to characterize the polarity and cell specificity of respiratory syncytial virus [26] and HPIV type 3 (HPIV3) [25], confirming that it is suited to studying paramyxovirus-HAE interactions that reflect those in the human lung.
We used viruses bearing HNs that are altered in receptor binding or F triggering to reveal the functional relevance of these properties in the HAE and to establish the key role of HN binding site II in infection in the natural host. We propose that an enhanced triggering of F by HN may be a disadvantage in vivo and that the function and timing of F triggering are critical in the target tissue. The correct balance between the three functions of HN (receptor binding, receptor cleaving, and F triggering) resides in the functions of the two binding sites [18], binding and release in site I and F triggering in site II, and both sites of HN play key roles in the natural host.
MATERIALS AND METHODS
HAE cell cultures
The EpiAirway AIR-100 system (MatTek Corporation) consists of normal human-derived tracheobronchial epithelial cells that have been cultured to form a pseudostratified, highly differentiated mucociliary epithelium closely resembling that of epithelial tissue in vivo. Upon receipt from the manufacturer, HAE cultures were transferred into six-well plates (containing 0.9 ml medium per well), with the apical surface remaining exposed to air, and incubated at 37°C in 5% CO2 overnight.
Monolayer cells
CV1 (African green monkey kidney) cells were grown in Dulbecco's modified Eagle's medium (Cellgro; Mediatech) supplemented with 10% fetal bovine serum and antibiotics at 37°C in 5% CO2.
Viruses
Titers of HPIV3 virus stocks were assessed by a plaque assay performed as described previously. [14] Variant HPIV3 strains were selected and grown as described previously. [13]
Infection and treatment of HAE cells and measurement of viral titers from infected HAE cells
HAE cultures were infected by applying 200 µl of EpiAirway medium containing 4,000 PFU of wild-type (wt) or variant HPIV3 to the apical surface for 90 min at 37°C. At 90 min, the medium containing the inoculum was removed, and cultures were placed at 37°C and fed each day with 0.9 ml medium via the basolateral surface. Viruses were harvested by adding 200 µl medium per well (with or without added treatments) to the HAE cultures' apical surface and allowed to equilibrate for 30 min at 37°C. The suspension was then collected, and viral titers were determined as previously described. [14] This viral collection was performed sequentially on the same wells of cells on each day postinfection. Treatments were performed by adding medium containing either 5 mM zanamivir or 0.5 units Clostridium perfringens neuraminidase (catalog number N2876; Sigma), and mixtures were diluted 150- to 10,000-fold during determinations of titers to eliminate any effect of the compounds on the subsequent titer infection. For experiments involving pH effects, CO2-independent medium was added during collection as described above at either pH 5.0 or pH 7.0. Harvesting by apical-surface washes (with or without treatment) was done sequentially on the same HAE cultures at 24-h intervals for the first 72 h and then at the indicated time points.
Immunoblotting
Twenty microliters of the fluid collected from the apical surface of HAE cells was subjected to immunoprecipitation with a mixture of two anti-HPIV3 HN monoclonal antibodies (MAbs) (77/5 and 170/7), supplied by Judy Beeler from the WHO repository, or with a mixture of anti-HPIV3 F MAbs (anti-F 108, supplied by Judy Beeler from the WHO repository, and mAb10207, from Chemicon) at a 1:100 dilution in a final volume of 50 µl. The samples were then resolved by 4 to 20% sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride membrane by electroblotting. The membranes were immunoblotted with polyclonal anti-HPIV3 antibodies (Cambrex) in 3% bovine serum albumin in phosphate-buffered saline (PBS) (1:1,000 dilution) and incubated with peroxidase-conjugated anti-guinea pig immunoglobulin G (which recognizes all HPIV3 proteins) using a 1:5,000 dilution. Tetramethylbenzidine (Promega) was added as described previously [9], and the images from the stained membranes were acquired using a Kodak 2000 multimodal image station and analyzed using Kodak molecular imaging software, version 4.0.
Immunofluorescent microscopy of infected HAE cells
HAE tissues were fixed in 4% paraformaldehyde-PBS and then frozen in OCT compound (Tissue-Tek). Frozen serial sections (5 µm) were obtained using a Leica CM3050 cryostat and mounted directly onto glass slides. Sections were air dried overnight, rehydrated, blocked, and immunostained using anti-HPIV3 antiserum (Alexander Schmidt, NIAID) at a 1:100 dilution in PBS, followed by detection with fluorescein isothiocyanate (FITC)-labeled goat anti-rabbit antiserum at a 1:1,000 dilution (Southern Biotech). Slides were mounted with Vectashield containing DAPI (4',6'-diamidino-2-phenylindole) and visualized with a fluorescence microscope (Nikon Eclipse E800) coupled to a Retiga digital camera (QImaging) and IPLab, version 3.65a, imaging software (Scanalytics).
Animals
Inbred cotton rats were obtained from Harlan (Indianapolis, IN). Female animals, 6 to 10 weeks of age, were used. The animals were purchased specific pathogen free according to the breeder's specification and were maintained in a barrier system. Animals were kept under controlled environmental conditions of 22 ± 1°C and a 12-hour light cycle. All animals were euthanized by CO2 inhalation.
Infection, virus titration, and histology.
For intranasal (i.n.) infection, HPIV3 was given in PBS to isoflurane-anesthetized cotton rats. i.n. inoculations of virus were administered in a volume of not more than 100 µl. Animals were asphyxiated using CO2, and lungs were removed and weighed. The left lung lobes were minced with scissors and dounced with a glass homogenizer, and the supernatant fluids were frozen until titration at –80°C. The right lung lobes were fixed by inflation with a 4% paraformaldehyde-PBS solution and paraffin embedded. The histological slides were stained with hematoxylin and eosin.
RESULTS
Viral growth in HAE cells is affected by HN's F-triggering properties and its receptor avidity. HAE cells cultured at an air-liquid interface [25, 26] form a highly differentiated epithelium that includes microvilli, cilia, and tight junctions; secretes mucin; and shows transepithelial resistance similar to that of in vivo tissue. These cells at 3 days of culture contain 48% ± 7% ciliated cells [5], closely mimicking the human trachea, in which ciliated cells account for approximately 50% of the lining. [4] Infection by wt recombinant HPIV3 in the HAE model system was shown to occur at the apical surface of ciliated epithelial cells and to be mediated by interactions with sialic acid residues. [25] To confirm these results in polarized cells for wt HPIV3, we tested the infection of both apical and basolateral surfaces of HAE cultures and found that progeny viruses are shed only from the apical surface of the HAE cells and that productive infection occurs only if the cells are infected apically (data not shown).

Table 1 To test the consequence of altered receptor avidity or F activation by the viral HN of HPIV3, we infected HAE cultures with viruses bearing mutations in receptor-binding site I (T193A) or site II (N551D and H552Q). Table 1 presents the relevant features of the variants used in the experiments described in this paper. The mutation at HN binding site I (T193A) confers the highest level of receptor-binding activity. [18] Single mutations at HN binding site II confer an enhanced fusion promotion capacity either with increased receptor avidity (H552Q) or with wt receptor avidity at a neutral pH but decreased avidity at a low pH (N551D). [15, 18] These mutations do not alter neuraminidase activity, which remains at wt levels for all variant HNs. These viruses have been well characterized in cultured cells [11] and allow us to investigate whether the HN mutants that efficiently trigger F, or avidly bind the receptor, have altered growth in the natural host epithelium. The HN variants, with their specific alterations in HN properties, provide a tool for dissecting HPIV3 pathogenesis and identifying specific determinants of infection in the lung.
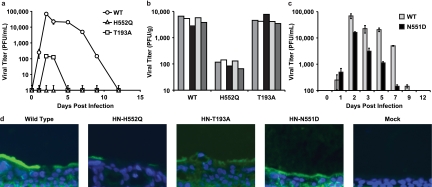
Figure 1 For the viral growth curves shown in Figure 1a, HAE cultures were infected via the apical surface, and viral production was assessed on the indicated days postinfection. We found that wt HPIV3 achieved the highest maximal titers in HAE cells. Interestingly, the site II variant (HN-H552Q), which bears an HN with a slightly higher receptor avidity than the wt and enhanced F triggering and is the most highly fusogenic in cultured monolayer cells [13], yields no detectable viral titers in HAE cells. The viral titer in HAE cells was also severely reduced by the mutation in site I (HN-T193A), which confers the highest receptor avidity (and is also highly fusogenic in monolayer cells) [13]; the plateau titers for this variant were approximately 3 logs below those for the wt virus. Thus, a series of mutations that alter HN functions and enhance cytopathicity in monolayer culture is actually detrimental to growth in HAE cultures.
The site I- and site II-mutated HN variant viruses grow poorly in the cotton rat
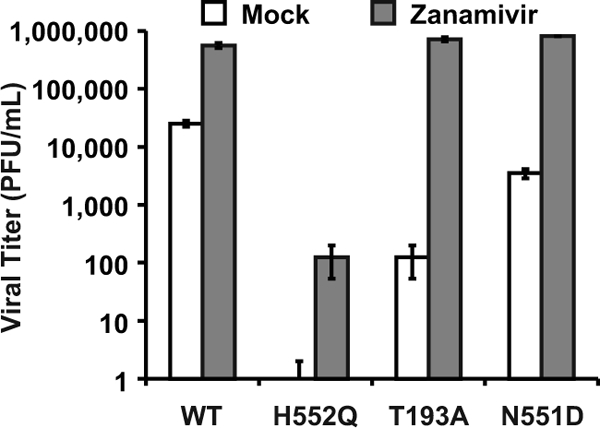
Figure 3 Paralleling the HAE culture results shown in Figure 1a are the animal data shown in Figure 1b, in which cotton rats were infected with wt or variant viruses. In this experiment, cotton rats were infected i.n. with 106.5 PFU of wt or variant virus, and lung titers of virus were determined 3 days after infection; day 3 was chosen because previously reported data from us and others show that the lung titer of wt HPIV3 peaks at this time point in the cotton rat and then declines over the next 3 days. [23, 24] Lungs were also harvested at days 5 and 6 after infection for histological examination (data not shown), since the histopathology in HPIV3-infected cotton rats is most intense at these later time points. [23, 24] As in the HAE cells, the HN-H552Q (site II) variant replicates to a negligible degree in vivo. Despite its fusogenicity in monolayer culture, this site II mutation is severely detrimental to growth in the animal. These results are consistent with our previously reported finding of severely curtailed nasal titers of the HN-H552Q variant in the cotton rat. [24] However, the HN-T193A variant grows at levels similar to those of the wt virus in the cotton rat. We propose that this finding is explained by the low pH in the lungs (pH ~6.5), which allows for more-efficient release from infected cell surfaces, even of a high-avidity variant, as we will explain in detail in connection with Figure 3a below.
HN's F-triggering efficacy independently modulates growth in the airway epithelium
In light of the finding that mutations in both sites I and II affect growth in HAE cells, we investigated a viral variant with a single mutation at HN binding site II (HN-N551D). The variant virus bearing HN-N551D (Table 1) was extensively characterized previously in cell culture and in biochemical assays for receptor binding, F-protein activation, and neuraminidase activity, and mutant HN was shown to be more efficient at triggering HPIV3 F despite wt neuraminidase activity and no increase in receptor avidity at a neutral pH. [15, 18] In monolayer cultures, HN-N551D grows and spreads to wt levels or even more efficiently than wt HPIV3. [15, 18] Figure 1c shows, however, that in HAE cells, this variant virus with wt receptor avidity but enhanced F triggering does not grow as well as wt HPIV3. However, this variant grows more efficiently than the two variants bearing HNs with high receptor avidity (compare to Fig. 1a). Thus, HN's F-triggering property has a separate impact on growth potential in the epithelium. Figure 1d shows infection of HAE cells with wt HPIV3 and each of the variant viruses viewed in histological cross section. The representative fluorescent photomicrographs localize viral antigens (FITC [green]); nuclei are stained with DAPI (blue). The cells were infected as described in the legend of Fig. 1a and c (or mock infected), incubated for 5 days, and stained with a polyclonal antibody specific for HPIV3. The viral antigens were detected, as expected, on the apical surface of HAE cells for wt HPIV3 and all variants. The differences in viral antigen detected between the variants rank in the order of the titers of infectious particles in Fig. 1a and c, with the exception of the HN-N551D variant (with enhanced F triggering), which was detected at or above wt levels on HAE apical surfaces despite releasing fewer infectious viruses than the wt. However, note that while the HN-H552Q variant (with enhanced F triggering and high receptor avidity [site II]) produced no infectious virus, there is clearly some viral antigen present at the HAE surface. Interestingly, for the HN-T193A (high receptor avidity [site I]) variant, the level of viral antigen at the HAE surface is only slightly lower than that of the wt despite the fact that no infectious particles could be detected at day 5 after infection.
The impaired viral production for the HN variant viruses, and the differences in the relationship between viral antigen on the HAE surface and infectious titer, could be explained by defects in either binding, entry, replication, or release, and the contribution of each property is discussed below.
Release of infectious particles is impaired for the site I/site II HN variants:
high receptor avidity hinders the subsequent progress of infection
The finding that viruses with high HN receptor affinity grow to low titers in HAE cultures suggests that the increased avidity for cell surface molecules may block the release of progeny virions from their aggregates on the lung cells' surface. We have previously shown that neuraminidase-deficient HPIV3 variants [6, 19] grow to low viral titers in monolayer cultures as a result of viral aggregation at the cell surface. The progeny virion's HN attaches to uncleaved receptor moieties on the host cell membrane or the envelope of other virions, preventing its release; this aggregation can be prevented by the addition of exogenous neuraminidase. [6, 19] The balance between HN's receptor-cleaving and receptor-binding functions appears to be key to effective virion release. Therefore, we next asked whether increased HN receptor avidity in the face of unchanged (wt) neuraminidase activity, as for the variants HN-T193A and HN-H552Q, may prevent the release of virions from the HAE surface. These two previously characterized fusogenic variant viruses have wt F and single mutations in HN: one with a mutation in a residue in the dimer interface region (H552Q) [13] and one with a mutation in the primary HN-binding site (T193A). [13] The T193A mutation confers increased receptor avidity in the face of wt neuraminidase and wt F activation activities; the H552Q mutation confers the highest of the variant receptor avidities, and also enhanced F-triggering function, in the face of wt neuraminidase activity (Table 1).
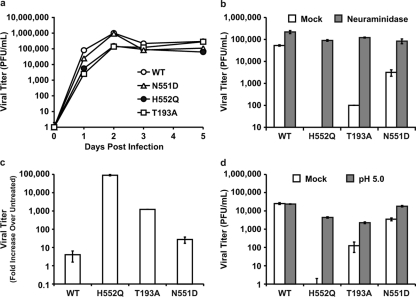
Figure 2 The experiment depicted in Figure 2a shows that supplemental neuraminidase treatment after infection restores wt viral titers for all three HN variants tested: HN-T193A (high avidity, site I), HN-H552Q (high avidity, site II), and also HN-N551D (wt avidity but enhanced F triggering, site II). Since these variants possess wt neuraminidase activity but altered avidity and F activation properties, these results reveal that the balance between HN's functions in receptor binding and receptor cleavage is important for growth in HAE cells. Note that viral entry is not impaired by the site I/site II mutations: with the addition of neuraminidase 30 min prior to the collection of the supernatant fluids, the titers are restored to wt levels, and thus, none of the viruses suffer defects in entry. Figure 2b compares the titers of virus on day 3 after infection of HAE cells with the addition of exogenous neuraminidase to those without the addition of exogenous neuraminidase and shows that each of the variants, despite very different titers without neuraminidase supplementation, attain wt titers upon supplementation. Figure 2c depicts the neuraminidase-induced increase in titer for each variant as a change over untreated controls and contrasts the magnitude of the correctible defects between HN mutants. On day 3, while the release of infectious PFU is enhanced 9-fold for the wt virus, the HN-T193A variant is enhanced 36-fold, and the HN-N551D variant is enhanced 24-fold. For the HN-H552Q variant, with high receptor avidity but wt neuraminidase activity, the release of infectious particles by neuraminidase supplementation is increased a striking 100,000-fold.
After finding that neuraminidase supplementation restores wt-level viral titers, we next tested a parallel strategy that avoids the addition of exogenous enzymes. Treatment under lower-pH conditions (closer to the pH optimum of the viral neuraminidase) at the time of viral harvest can be used to enhance the activity of the viral neuraminidase. [20] Each of the different HNs has wt neuraminidase activity; the increase induced by lowering the pH is equivalent for all of them and has been shown to enhance the release of HN from receptor-bearing target cells. [15, 20] The use of low pH and optimization of viral neuraminidase, rather than exogenous neuraminidase, ensure that the specific sialic acid receptors for the virus are cleaved rather than the diverse array of sialic acid-containing molecules that are removed by bacterial neuraminidase. The experiment shown in Figure 2d shows that the optimization of neuraminidase activity under low-pH conditions (pH 5.0) enhanced the release of PFU by both site II variants HN-H552Q and HN-N551D and site I variant HN-T193A.
These results show that the failure of budded virions to be released from the cell surface contributes to the low titers of high-affinity HN variants; however, these defects in release cannot be explained by avidity differences alone. The receptor avidity of the T193A mutant HN is the highest of all the variants, substantially higher than that of the HN-H552Q [18], and yet the decrease in viral titer was less significant than that of the HN-H552Q variant (Fig. 1a). The effect of neuraminidase on virus release did not correlate directly with receptor avidity; the greatest increase in the release of virus with neuraminidase treatment was seen for the HN site II variant with lower avidity (HN-H552Q) rather than the HN variant with the highest avidity (HN-T193A) (site I) (Fig. 2). These results suggested that viral release from the infected cell is modulated not only by receptor avidity but also by additional factors. We next investigated the role of HN's F-triggering function in the viral life cycle in HAE cells.
Dysregulation of F triggering by mutations in HN site II impairs viral growth in HAE cells. While the site II mutation that increases receptor avidity (HN-H552Q) led to the greatest impairment in the release of infectious viral particles shown in Fig. 1, the site II mutation that enhances F triggering without affecting receptor avidity at neutral pH (HN-N551D) was also impaired in release. We propose that the alteration in HN's F-triggering function caused by HN site II mutations contributes to the decreased viral titer observed in the experiments shown in Fig. 1.
During viral exit from the host cell, viral neuraminidase cleaves HN's receptor, thus preventing both sites I and II from interacting with the receptor and allowing the release of progeny virions. The receptor analog zanamivir provides a different method of enhancing viral release by blocking site I receptor binding while not affecting site II function. [18, 21] Therefore, zanamivir, by competing with sialic acid receptors for HN's binding site I, also prevents virion aggregation and can release cell-bound virions. [19] The experiment in Figure 3 shows that the treatment of infected HAE cultures with zanamivir increased the viral titers of the HN-N551D variant (which has enhanced F triggering and wt receptor avidity) by 2 logs. Zanamivir treatment also enhanced the growth (titer) of wt virus and the growth of the two variants bearing high-avidity HNs. The titers for wt virus increased by more than 1 log, and variant HN-T193A (site I) now attained wt levels, with an increase of 4 logs. The infectious viral titers for HN-H552Q (site II mutant), however, which were undetectable without treatment, rose by 2 logs but were still 6,000-fold lower than wt titers and failed to attain wt titers in HAE cultures even in the presence of zanamivir. The likely explanation for this finding lies in the fact that zanamivir interacts only with site I and not with site II. [18, 21] We proposed that site II could mediate persistent binding to the cell surface or that an unbound site II may retain its F-triggering potential. In the case of the HN-H552Q variant, whose site II has enhanced receptor avidity in addition to enhanced F triggering, this could lead to the efficient inactivation of new virions. We tested this hypothesis in the next set of experiments.
Dysregulation of F triggering by HN results in the production of noninfectious particles
The experiments depicted in Fig. 2 and 3 show the striking distinction between the effect of mutations in site I versus that of mutations in site II. Both zanamivir and neuraminidase raise the titers of high-avidity site I variants to wt levels (i.e., within 1 log of the wt titer), but only neuraminidase raises (to the wt level) the infectious titer of a site II mutant with enhanced F triggering (HN-H552Q). Therefore, we hypothesized that an enhanced action of site II is disadvantageous for the production of infectious virions. If this were true, then viral particles might be produced and released as noninfectious particles unless site II action is prevented by the elimination of its receptor (i.e., by neuraminidase treatment).
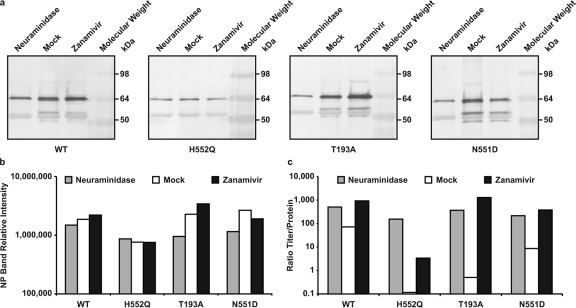
Figure 4 In the experiment shown in Figure 4, we tested this hypothesis by quantifying the amount of particles released by each variant (as viral protein) in response to each treatment and comparing this result to the infectious titer of the variants under each condition. For each supernatant fluid sample from the infected HAE cultures in Fig. 2 and 3 for which the titer was determined, a parallel sample was reserved for viral protein analysis. Equal amounts of each sample were immunoprecipitated with a mixture of anti-HN antibodies (see Materials and Methods), subjected to Western blot analysis, and probed with polyclonal anti-HPIV3 antibodies. The results of the immunoprecipitation, as shown in Figure 4a, reveal the differences in overall particle release for the wt and the variants HN-T193A, HN-H552Q, and HN-N551D.
Figure 4b shows the densitometric quantitation of the Western blot analysis shown in Fig. 4a for each variant and treatment. This quantitation was performed on the nucleoprotein (NP) band since this protein is predominant and offers an accurate comparison. For the wt and all HN variants, neuraminidase treatment results not only in similar numbers of PFU (as seen in Figure 2) but also in similar viral protein intensities (Fig. 4b). Zanamivir treatment (Fig. 4b) of HAE cells infected with the HN-N551D variant, which increased the titer for this variant by 200-fold compared to that of mock treatment (Fig 2), shows no corresponding effect on viral protein intensity; the amount of HN-N551D variant viral protein is similar after mock or zanamivir treatment. For HN-H552Q, although the titer in the presence of mock treatment was virtually zero (undetectable PFU) (Fig. 2) and was enhanced by zanamivir, the amount of viral protein after mock treatment is similar to that after zanamivir treatment (Fig. 2). Similar results were obtained with the immunoprecipitation performed using anti-F MAbs (see Materials and Methods) (data not shown).
DISCUSSION
Regulation of fusion during the paramyxoviral life cycle in airway cells.
The growths of wt HPIV3 and variant viruses in the HAE cultures closely reflect those in the cotton rat lung and thus represent an excellent system for testing hypotheses regarding the interplay of the host cell and viral factors in pathogenesis. While HPIV3 and variants induce syncytium formation in cultured monolayer cells [6, 13, 18], no cell-cell fusion was observed in HAE cells (data not shown), as observed previously. [25] The infection of cotton rats with wt or variant HPIV3 in our experiments (Figure 1) also did not result in any cell-cell fusion (data not shown), further reflecting the similarity between these two systems. Using HPIV3-infected HAE cells, Zhang et al. [25] previously showed that the F protein is trafficked exclusively to the apical surface of ciliated cells, suggesting that this localization results in a limited interaction of F with neighboring cells that may account for the lack of syncytium formation. Consistent with this idea, despite the extensive cultured cell fusion that is the hallmark of several of the HN variants [6, 13, 18], no cell-cell fusion was observed in infected HAE cells using either light microscopy or electron microscopy (data not shown).
The correct timing of F activation (triggering) is essential for entry; for infection, triggering must occur only when F is in proximity to the target cell membrane. The finding that HN's F-triggering feature, in addition to its receptor avidity, has functional relevance in a model that mimics the natural host represents an important new paradigm to explain paramyxovirus virulence and pathogenesis. The results suggest that, indeed, the dysregulation of F triggering prohibits the successful completion of the viral life cycle and indicate that strategies to perturb the timing of F triggering will have a biological impact in vivo.
Relevance of HN's F activation activity to in vivo growth.
Differences between viruses that bear variant HNs provide the key to our interpretations in this study. While the HN site II-mutated variant HN-H552Q is extremely fusogenic in monolayer cultures, it did not replicate in the cotton rat, leading to the hypothesis that enhanced triggering may be a disadvantage in vivo. We tested this hypothesis in our model HAE culture and indeed found that HN site II mutants (N551D and H552Q) have a disadvantage in HAE culture that cannot be explained simply by increased receptor avidity. We have shown that the triggering-enhanced (HN-H552Q) viruses are in fact budding from the surface of the airway epithelial cells but are mostly noninfectious. This could be due to either virus aggregation or inactivation, e.g., by premature F triggering. However, efforts to dissociate potentially aggregated particles in the collected supernatant fluid samples with neuraminidase (data not shown) did not yield higher titers of infectious virus, and therefore, we favor the hypothesis that the particles are released but are not infectious.
Relevance of HN's receptor avidity to in vivo growth.
While adequate receptor avidity is essential for viral entry, we have shown that it is the balance between the features of receptor binding, receptor cleavage, and F triggering that modulates infection. [20] The HAE model system reveals that the wt virus possesses the optimal balance of these properties. Our results indicate that higher-avidity HN molecules result in viruses that are defective in release from the infected cell or that release noninfectious particles. The effect of receptor avidity is highlighted by the interesting variant with an HN site II mutation that confers altered F triggering but wt avidity at pH 7.0 (HN-N551D). For this particular site II variant, lowering the pH decreases the receptor avidity because the N551D mutation confers pH sensitivity, i.e., lower avidity at pH 5.0 than at pH 7.0. [15] The fact that lowering the pH to 5.0 enhances release of this virus reflects the inverse relationship between receptor avidity and infectious viral titer yielded by HAE cultures. The release of infectious particles is key for the disease process: only upon the release of progeny virions after the initial round of infection can virus spread to new host cells, leading to the establishment of ongoing infection and disease. These results are of particular interest since the human airway surface is mildly acidic in pH, with an average pH value of 6.6 in healthy individuals. During bacterial infection, the endobronchial pH can decrease significantly, to values between pH 5.6 and 6.2, and in patients with cystic fibrosis, the airway pH is also lower than normal, around pH 6.0. [4] Our results suggest that acidic conditions in the airway could increase the production of infectious virus during HPIV3 infection, permitting natural variant viruses as well as wt viruses to persist and, potentially, thus enhance the spread of virus within the lung and the establishment of disease.
Implications for antiviral development.
No clinical therapies or vaccines exist for parainfluenza viruses, and vaccines would be unlikely to protect the most-vulnerable youngest infants. The development of specific antiviral agents is therefore important. The results presented here reveal that the HAE model replicates the pathogenesis of HPIV3 data in cotton rats, implying that specific features of viral entry and release that affect pathogenesis can be modeled in this system. This means that the HAE model can be used to screen for inhibitory molecules that would be effective in vivo.
One attractive antiviral strategy for paramyxoviruses is the use of fusion peptide inhibitors that interfere with the conformational changes in the F protein that occur after activation and that lead to a membrane merger during viral entry. [17] Properties of HN that modulate the rate or efficiency of F activation affect the antiviral efficacy of peptide inhibitors in vitro. [22] The results here for HAE cells imply that these effects will apply in vivo as well and suggest that variants that escape peptide inhibition via alterations in F triggering may have a growth disadvantage in the natural host.
Perhaps the most direct strategy for preventing infection is by interfering with the initial receptor binding, either by depleting or blocking sialic acid-containing receptors or by competitively inhibiting HN's receptor-binding sites. [11] For influenza virus, a recombinant fusion protein consisting of the neuraminidase catalytic domain of Actinomyces viscosus fused with a cell surface-anchoring sequence (Fludase; DAS181) removes sialic acid-containing receptors for influenza virus from the airway epithelium. This compound is an effective inhibitor of influenza virus infection in vitro and in animal models. [8, 12] Given the similarities in the initial binding receptors used by HPIV and influenza virus [1], this strategy may also be attractive for HPIV. In fact, Zhang et al. showed that HAE cells treated with Vibrio cholerae neuraminidase were not infectible by HPIV for 24 h. [25] The HN variant viruses discussed in this paper, mutated in site I or site II of HN, emerged under the pressure of the neuraminidase-mediated depletion of receptors in cell monolayers. The variants were those that escaped the constraint of receptor depletion either by enhanced receptor avidity or by enhanced F triggering. Thus, the concern might be raised that such resistant variants might emerge under the selective pressure of drug treatment in vivo. However, the results presented here show that in the natural host epithelia, the features that confer resistance to receptor depletion also confer severe growth disadvantages. In the case of influenza virus, decreased receptor availability might be countered by a more-avid receptor-binding molecule to facilitate viral entry in the face of receptor scarcity. Thus, influenza viruses might escape such an antiviral compound by evolving a higher-avidity hemagglutinin molecule. However, for HPIV3, our results indicate that a more-avid HN would result in defective release from the infected cell or in release of noninfectious particles, and therefore, this selective pressure would likely not yield a fit, transmissible, resistant virus.
In light of the finding that the dysregulation of F triggering prohibits successful infection, our results also suggest that antiviral strategies designed to perturb the timing of F triggering have promise. The finding that mutant HN-H552Q, with an HN that likely leads to a prematurely activated F, grows poorly and produces noninfectious progeny particles indicates that dysregulating F triggering is an attractive antiviral strategy. While F activation is key for entry, the correct timing of this activation is essential; triggering must occur when F is in contact with the target cell membrane. We suggest that this timing of activation represents a potential target for intervention [20], and the results presented here support the notion that HN's F triggering represents a potential target. We contend that HN molecules with alterations at site II that emerge under the selective pressure of treatment with such drugs would be impaired in growth in the airway, as for the site II variants which we discuss here. If efficient receptor mimics can trigger and thus inactivate F after budding and release, then released virus could be rendered noninfectious, and viral spread could be halted. This is of clinical relevance since therapy for respiratory viral disease will generally need to be effective when used after the onset of infection.
ACKNOWLEDGMENTS
This work was supported by Public Health Service grant AI31971 from the National Institutes of Health (NIAID), a Gates (Grand Challenge) phase I award, and a March of Dimes research grant to A.M. and by an American Lung Association research grant to M.P. L.M.P. is supported by a Parker B. Francis Fellowship in Pulmonary Research and a Stony Wold-Herbert Fund, Inc., fellowship.
We are grateful to Ashton Kutcher, Jonathan Ledecky, and Nikon, Inc., for support of our microscopy; to Dan and Nancy Paduano for support of innovative research projects; and to the Friedman Family Foundation for renovation of our laboratories at Weill Cornell Medical College. We thank Hang-Sun Kim for experimental assistance, the Shaklee Corporation for support of our airway epithelial tissue model, Ray Pickles for helpful discussions, Alexander C. Schmidt (Laboratory of Infectious Diseases, NIAID) for the gift of anti-HPIV3 antibodies for immunofluorescent microscopy, and Judy Beeler (WHO repository) for anti-HPIV3 HN and anti-HPIV3 F antibodies.
References:
Ah-Tye, C., S. Schwartz, K. Huberman, E. Carlin, and A. Moscona. 1999.
Virus-receptor interactions of human parainfluenza viruses types 1, 2 and 3.
Microb. Pathog. 27329-336Corey, E. A., A. M. Mirza, E. Levandowsky, and R. M. Iorio. 2003.
Fusion deficiency induced by mutations at the dimer interface in the Newcastle disease virus hemagglutinin-neuraminidase is due to a temperature-dependent defect in receptor binding.
J. Virol. 776913-6922Deng, R., Z. Wang, P. J. Mahon, M. Marinello, A. Mirza, and R. M. Iorio. 1999.
Mutations in the Newcastle disease virus hemagglutinin-neuraminidase protein that interfere with its ability to interact with the homologous F protein in the promotion of fusion.
Virology 25343-54Fischer, H., and J. H. Widdicombe. 2006.
Mechanisms of acid and base secretion by the airway epithelium.
J. Membr. Biol. 211139-150Gomperts, B. N., L. J. Kim, S. A. Flaherty, and B. P. Hackett. 2007.
IL-13 regulates cilia loss and foxj1 expression in human airway epithelium.
Am. J. Respir. Cell Mol. Biol. 37339-346Huberman, K., R. Peluso, and A. Moscona. 1995.
The hemagglutinin-neuraminidase of human parainfluenza virus type 3: role of the neuraminidase in the viral life cycle.
Virology 214294-300Li, J., V. R. Melanson, A. M. Mirza, and R. M. Iorio. 2005.
Decreased dependence on receptor recognition for the fusion promotion activity of L289A-mutated Newcastle disease virus fusion protein correlates with a monoclonal antibody-detected conformational change.
J. Virol. 791180-1190Malakhov, M. P., L. M. Aschenbrenner, D. F. Smee, M. K. Wandersee, R. W. Sidwell, L. V. Gubareva, V. P. Mishin, F. G. Hayden, D. H. Kim, A. Ing, E. R. Campbell, M. Yu, and F. Fang. 2006.
Sialidase fusion protein as a novel broad-spectrum inhibitor of influenza virus infection.
Antimicrob. Agents Chemother. 501470-1479McKimm-Breschkin, J. L. 1990.
The use of tetramethylbenzidine for solid phase immunoassays.
J. Immunol. Methods 135277-280Melanson, V. R., and R. M. Iorio. 2004.
Amino acid substitutions in the F-specific domain in the stalk of the Newcastle disease virus HN protein modulate fusion and interfere with its interaction with the F protein.
J. Virol. 7813053-13061Moscona, A. 2005.
Entry of parainfluenza virus into cells as a target for interrupting childhood respiratory disease.
J. Clin. Investig. 1151688-1698Moscona, A. 2008.
Medical management of influenza infection.
Annu. Rev. Med. 59397-413Moscona, A., and R. W. Peluso. 1993.
Relative affinity of the human parainfluenza virus 3 hemagglutinin-neuraminidase for sialic acid correlates with virus-induced fusion activity.
J. Virol. 676463-6468Murrell, M. T., O. Greengard, M. Porotto, N. Poltoratskaia, and A. Moscona. 2001.
A single amino acid alteration in the human parainfluenza virus type 3 hemagglutinin-neuraminidase glycoprotein confers resistance to the inhibitory effects of zanamivir on receptor binding and neuraminidase activity.
J. Virol. 756310-6320Palermo, L. M., M. Porotto, O. Greengard, and A. Moscona. 2007.
Fusion promotion by a paramyxovirus hemagglutinin neuraminidase protein: pH modulation of receptor avidity of binding sites I and II.
J. Virol. 819152-9161Pickles, R. J., D. McCarty, H. Matsui, P. J. Hart, S. H. Randell, and R. C. Boucher. 1998.
Limited entry of adenovirus vectors into well-differentiated airway epithelium is responsible for inefficient gene transfer.
J. Virol. 726014-6023Porotto, M., P. Carta, Y. Deng, G. Kellogg, M. Whitt, M. Lu, B. Mungall, and A. Moscona. 2007.
Molecular determinants of antiviral potency of paramyxovirus entry inhibitors.
J. Virol. 8110567-10574Porotto, M., M. Fornabaio, G. E. Kellogg, and A. Moscona. 2007.
A second receptor binding site on human parainfluenza type 3 hemagglutinin-neuraminidase contributes to activation of the fusion mechanism.
J. Virol. 813216-3228Porotto, M., O. Greengard, N. Poltoratskaia, M.-A. Horga, and A. Moscona. 2001.
Human parainfluenza virus type 3 HN-receptor interaction: the effect of 4-guanidino-Neu5Ac2en on a neuraminidase-deficient variant.
J. Virol. 767481-7488Porotto, M., M. Murrell, O. Greengard, L. Doctor, and A. Moscona. 2005.
Influence of the human parainfluenza virus 3 attachment protein's neuraminidase activity on its capacity to activate the fusion protein.
J. Virol. 792383-2392Porotto, M., M. Murrell, O. Greengard, M. Lawrence, J. McKimm-Breschkin, and A. Moscona. 2004.
Inhibition of parainfluenza type 3 and Newcastle disease virus hemagglutinin-neuraminidase receptor binding: effect of receptor avidity and steric hindrance at the inhibitor binding sites.
J. Virol. 7813911-13919Porotto, M., C. C. Yokoyama, G. Orefice, H.-S. Kim, M. Aljofan, B. A. Mungall, and A. Moscona. 2009.
Kinetic dependence of paramyxovirus entry inhibition.
J. Virol. 836947-6951Porter, D., G. Prince, V. Hemming, and H. Porter. 1991.
Pathogenesis of human parainfluenza virus 3 infection in two species of cotton rat: Sigmodon hispidus develops bronchiolitis, while Sigmodon fulviventer develops interstitial pneumonia.
J. Virol. 65103-111Prince, G. A., M. G. Ottolini, and A. Moscona. 2001.
Contribution of the human parainfluenza virus type 3 HN-receptor interaction to pathogenesis in vivo.
J. Virol. 7512446-12451Zhang, L., A. Bukreyev, C. I. Thompson, B. Watson, M. E. Peeples, P. L. Collins, and R. J. Pickles. 2005.
Infection of ciliated cells by human parainfluenza virus type 3 in an in vitro model of human airway epithelium.
J. Virol. 791113-1124Zhang, L., M. E. Peeples, R. C. Boucher, P. L. Collins, and R. J. Pickles. 2002.
Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology.
J. Virol. 765654-5666
Return to SHAKLEE STUDIES
Since 9–09–2017
| Home Page | Visit Our Sponsors | Become a Sponsor |
Please read our DISCLAIMER |