

The Nosological Classification of
Whiplash-associated Disorder:
A Narrative ReviewThis section is compiled by Frank M. Painter, D.C.
Send all comments or additions to: Frankp@chiro.org


FROM: J Can Chiropr Assoc 2021 (Apr); 65 (1): 76–93 ~ FULL TEXT
OPEN ACCESS Joe H. Ghorayeb, DC, MHA, DESS
University of Medicine and Health Sciences,
New York, NY.
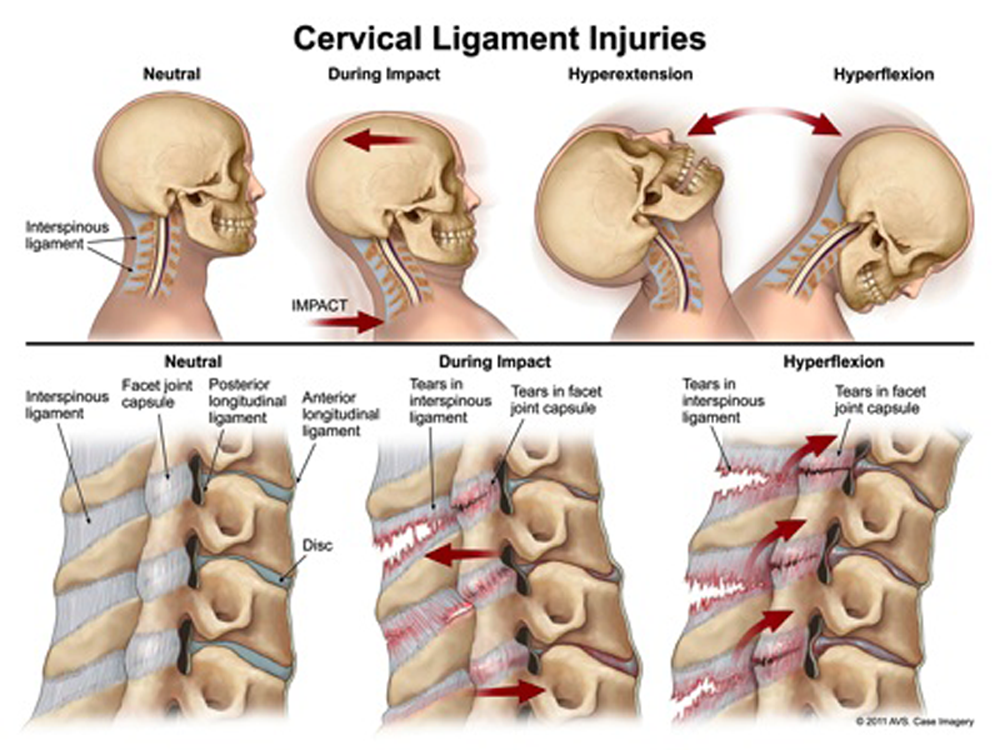
Whiplash-associated disorder (WAD) is the most common complaint and purported cause of chronic disability associated with motor vehicle collisions in North America. However, its construct validity remains controversial. This narrative review of the literature summarises the evidence underlying the most commonly theorised biological and psychosocial mechanisms of WAD pathogenesis. While the face validity of WAD is good, empirical evidence supporting the various constructs suggesting a causal link between a trauma mechanism and the development of symptoms is poor. Because individual expectations of recovery are outcome-predictive, future research is necessary to develop a better understanding of how to enhance expectancies in order to help affected motorists gain a greater sense of control over their health and wellbeing.
Keywords: chiropractic; expectancies; injury; nosology; pathology; whiplash.
The FULL TEXT Article:
Introduction
Whiplash-associated disorder (WAD) is the most commonly reported clinical presentation by individuals involved in motor vehicle collisions (MVCs) with an annual incidence that has been estimated to be 300 per 100,000 people in North America. [1] The term “whiplash” was first introduced by Crowe in 1928 when he described the motion of sudden acceleration-deceleration of the cervical spine as the result of a collision. [2] Crowe did not intend for the expression to reflect the name of a new disease. [3] In spite of this goal, clinicians, patients and lawyers have accepted whiplash as a clinical entity with a wide variety of sequalae, purportedly caused by trauma to the spine and its surrounding structures.
The Quebec Task Force on Whiplash Associated Disorders classifies WAD based upon the severity of a person’s symptoms and signs: [4]
Grade 0: No complaints about neck pain. No physical signs.
Grade I: Neck complaint of pain, stiffness or tenderness only. No physical signs.
Grade II: Neck complaint and musculoskeletal signs including decreased range of motion and point tenderness.
Grade III: Neck complaint, musculoskeletal signs and neurological signs including decreased or absent deep tendon reflexes, muscle weakness and sensory deficits.
Grade IV: Neck complaint and fracture or dislocation.
The clinical presentation of WAD is highly variable. In one study of 6,481 Saskatchewan residents who filed auto insurance claims, only 0.4% of respondent complaints were restricted to neck pain alone, with pain often extending into the head, shoulder girdle, upper, mid and lower back, and the upper and lower extremities. [5] A variety of psychological symptoms may also be associated with WAD, including depression, anger, fear, anxiety, and hypochondriasis. [6]
Several kinematic studies have attempted to glean more information with respect to the forces applied to the body during a collision that may be helpful in understanding the etiology of WAD and inform its management. Suggested sites of tissue injury include the facet joint, capsule and its ligaments [7, 8], intervertebral disc [9], spinal ligaments [10], and skeletal muscle. [11] Evidence also exists to suggest that central nervous system involvement [12], as well as psychosocial factors [13, 14] play a role in the development and persistence of WAD.
Given the pertinence of periodic review to revise our understanding of the scientific literature on the etiology of WAD, the aim of this narrative review is to summarise the evidence underlying the most commonly suggested mechanisms of WAD pathogenesis and to provide an updated appreciation of the subject matter in the 25 years since the last such review was performed by Stovner [15] as part of a treatise on the etiopathology of WAD.
This review also considers whether a causal link between a trauma mechanism and the development of symptoms is reasonable. This is accomplished through the widely accepted guidelines for causation, established by Sir Austin Bradford Hill [16], though not without controversy [17–19], to discern the fundamental prerequisites and assessment criteria of the cause-effect relationship as it relates to WAD pathogenesis.
Methods
Search strategy
A MEDLINE/PubMed search was performed using the following search string “whiplash [Title/Abstract} AND injury [Title/Abstract} AND pathogenesis [Title/Abstract]” from January 1, 1994 to December 31, 2019, yielding 1,237 entries, which were exported to EndNote X9 for reference management and tracking of the screening process.
Selection of studies
Studies were included if they met the following criteria:1) published in English and in a peer-reviewed journal;
2) study designs included experimental (Randomised Clinical Trials) and observational (Cohort and Case-Control) studies;
3) study populations include adults (19+ years old);
4) study populations confined to traffic collisions;
5) studies that suggest damage or injury to, or aberration of, commonly suspected anatomical structures of the body including:
- the facet joint and/or capsule,
- skeletal muscle, and
- the central nervous system;
6) studies that suggest psychological factors associated with WAD such as psychological distress, catastrophisation, and patient expectancies, and
7) studies that consider compensation and its role in health outcomes.

Table 1
Table 2
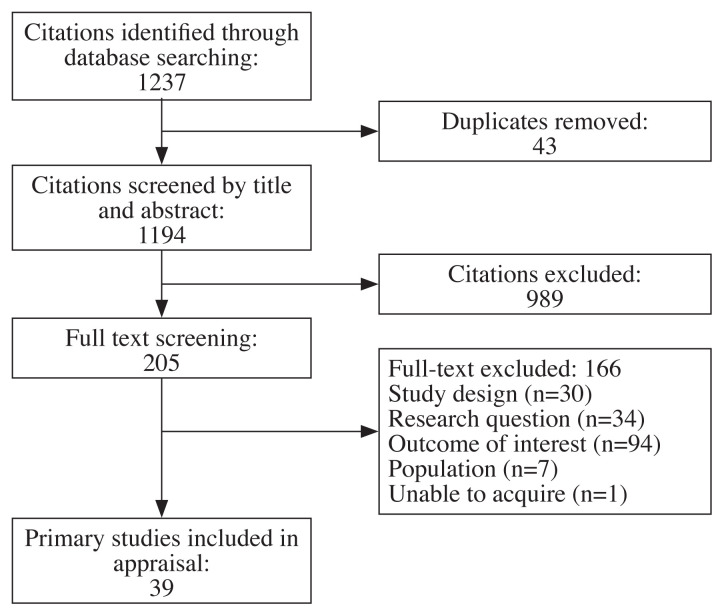
Figure 1
Table 3
Table 4
Table 5
Table 6
Table 7 Because limited evidence exists to substantiate the notion that intervertebral disc [20–22] and spinal ligament [23, 24] involvement materially contribute to the development of WAD, summary regarding the validity of these particular structures as potential causes of WAD was not included in this review.
Studies concerning grade IV WAD, the pediatric population, esophageal, ocular, oropharyngeal, otologic, temporomandibular joint or vascular manifestations that may be associated with the condition, as well as cadaveric studies, kinematic studies, case reports, opinions, comments, letters to the editor, and articles without scientific data or a report of their methodology were excluded.
Level of evidence
A level of evidence rating was given to every study, based on the study design (Table 1), according to the 2005 classification system of the Dutch Institute for Healthcare Improvement CBO. Randomised double-blinded comparative clinical research of good quality and efficient size obtained a level of evidence A2, while cohort studies not meeting these criteria or case-control studies obtained a level of evidence B. Non-controlled trials obtained a level of evidence C. The level of evidence for each study is listed in the corresponding table of studies suggesting a cause of WAD, and the overall level of evidence for the cluster of studies represented in each table is listed under the respective table.
Strength of conclusion
Subsequently, the strength of conclusion (ranging from 1 to 4) was calculated for each cluster of studies reflecting one outcome parameter (Table 2) and is placed under the respective table representing a cluster of studies suggesting a cause of WAD. Strength of conclusion 1 was assigned for a study of level A1 or at least 2 independently conducted studies of level A2. Strength of conclusion 2 was given when at least 2 independently conducted studies of evidence level B or one trial of evidence level A2 was included in the cluster, and strength of conclusion 3 was assigned if one study of evidence level B or C was present. Strength of conclusion 4 was given in case of inconclusive or inconsistent results between various studies.
The author reviewed all entries, of which, 43 duplicates were removed, and 1,194 citations were screened by title and abstract. Two hundred and five full-text articles were screened. Of those, 39 articles met the inclusion criteria and were eligible for critical appraisal. Reasons for exclusion during the full-text screening phase were study design (n=30), research question (n=34), outcome of interest (n=94), and population of subjects (n=7) (Figure 1).
The studies selected for further analysis were clustered according to the following theorised mechanisms of WAD pathogenesis:1) evidence of facet joint and/or capsule injury (Table 3);
2) evidence of change in skeletal muscle morphology (Table 4);
3) evidence of central nervous system involvement (Table 5);
4) evidence of a role of psychological distress and patient expectancies (Table 6); and
5) evidence of the role of compensation resulting in poor health outcomes (Table 7).
Results
Facet joint and capsule injury
Of the various tissues in the cervical spine that may be injured during whiplash, the facet (zygapophysial) joint and facet capsule/ligament are the most extensively documented structures. A proposed mechanism of facet joint injury is undo strain upon the facet capsule, which has been documented in several in vitro biomechanical, and in vivo animal, studies. [25–32] Given that such studies are not possible in live humans, attempts to infer injury to the facet joint have been made by way of eight clinical trials documenting pain relief when the facet joint and capsule are specifically targeted via medial branch blocks or percutaneous radiofrequency neurotomy with some meaningful pain relief achieved. Of these studies, four were randomised clinical trials [33–36]; three involved anesthetic block [33–35] and five radiofrequency neurotomy. [36, 38–41]
Two of the three RCTs involved WAD patients with headaches ± neck pain who underwent facet blocks. In two of these studies, patients were randomly assigned to receive an initial radiographically-guided block with either short-acting 2% lignocaine or longer-acting 0.5% bupivacaine. [33, 34] A positive diagnosis of cervical zygapophysial joint pain was made only if both blocks relieved a patient’s pain and bupivacaine provided longer relief, which occurred in 13 of 55 patients with headache complaints and 14 of 55 patients with neck pain. [33] The second study saw 27 of 50 patients obtain neck pain relief based on a similar protocol. [34] In the third study, patients who presented with dominant headache and obtained relief following third occipital nerves blocks satisfied the authors’ criteria for C2–C3 zygapophysial joint pain, and nonresponders along with patients who presented with dominant neck pain underwent blocks of cervical zygapophysial joints at several levels below C2–C3. [35] Responders were then diagnosed with cervical zygapophysial joint pain at the respective level that offered them relief. The major limitation of the aforementioned studies, pertaining to the current discussion, was the outcome of interest (headache relief vs neck pain relief) coupled with the heterogeneous sample of patients, suggesting a significant risk of selection bias.
The only parallel-group RCT [36] involved 24 patients presenting with what the authors deemed to be cervical zygapophyseal joint pain at levels C3–C7; confirmed with the use of local anesthetic blocks. Patients with C2–C3 zygapophyseal joint pain were excluded because a previous study [37] had shown that treatment at this level by radiofrequency neurotomy was technically difficult. Each subject received either active or mock radiofrequency neurotomy (n = 12 per group) and was followed by telephone interviews and clinic visits until they reported that their pain had returned to 50% of its preoperative level. The treatment protocol in the two arms was identical, except that the temperature of the electrode tip was raised to 80°C for 90 seconds in the active group and maintained at 37°C in the control group. Six patients in the control group and three in the active-treatment group had a return of their accustomed pain immediately following the procedure. At 27 weeks, one patient in the control group and seven in active-treatment group remained pain free with a median time of eight days and 263 days, respectively, before the return of at least 50% of their preoperative pain. [36] Notably, a higher proportion of patients involved in ongoing litigation were assigned to the control group. How this may have affected the study results remains unclear given the lack of information provided regarding these patients’ circumstances.
The remaining four clinical trials employing radiofrequency neurotomy were uncontrolled. [38–41] The aforementioned studies operate on the following assumption: if we accept that a patient reported no pain prior to an MVC but reports pain thereafter, which is successfully treated with facet blocks or radiofrequency neurotomy, the inference is that the facet joint is the source of the patient’s pain and, therefore, was injured or implicated in some way during the MVC. This is a common logical fallacy known as post hoc ergo propter hoc (from the Latin meaning: “with this, therefore because of this”) – where credit is awarded to the treatment administered rather than considering a condition’s natural history, regression to the mean, or the impact of narratives proffered to a patient, coupled with the power of the placebo effect and the contextual factors surrounding a patient’s particular circumstances that work in concert to bring about recovery.
High-quality evidence demonstrates significant variability in the diagnostic utility of cervical facet joint nerve blocks in individuals with chronic spinal pain; with prevalence rates ranging between 36% to 67% and false-positive rates of 27% to 63%. [42] Moreover, because studies of healthy and clinical populations find little difference in the prevalence of anatomical impairment to warrant procedural intervention [43–46], in turn, challenging the practice of assigning a structural cause for the perception of pain in the absence of detectable tissue pathology, an appreciation for the neurobiology of persistent pain is recommended. [47]
In the context of WAD, it is noteworthy that the reporting of symptoms is not synonymous with the presence of injury, which refers to damage to the body produced by energy exchanges that have relatively sudden discernible effects. [48] Additionally, the perception of pain may arise from an emotional experience [49], as the correlation between pain and bodily damage is equivocal.
Although treatment interventions targeted at the facet joint have been shown to provide symptomatic relief for a small cohort of patients, the conclusion that injury to the facet joint is therefore the cause of WAD cannot be drawn ipso facto.
Muscle involvement
Muscle pain is a prevailing symptom reported by motorists involved in traffic crashes. Though, evidence of direct muscle injury as a cause of WAD remains inconclusive. [50] Several studies have documented findings in neck muscles by ultrasonography, including muscle deformation during real-time movement [51, 52], muscle twitching [53], and the temporal development of fatty infiltration of the multifidus following MVC exposure. [54–57] In a single-center longitudinal cohort study by Elliot and colleagues [54], 36 people with acute WAD (≤1 week) were followed and assessed for temporal development of muscle fatty infiltrates (MFI) in the cervical multifidii and the findings were measured against self-reported pain and disability via the Neck Disability Index (NDI). Study subjects were dichotomised into two groups via NDI (0–28%, recovered/ mild disability and 30–100%, moderate to severe disability). Mean percent MFI by group and time revealed little variation in the within group changes in the recovered group, the modest mean changes between one week and three months were statistically significant (p=0.023). In the moderate/severe disability group, mean percent MFI significantly increased across all time points (p<0.002). Comparing the recovered/mild to moderate/severe groups indicated no significant difference at 1 week (p=0.31) with significant differences at two weeks (p=0.0009) and at three months (p<0.0001). Although underpowered, this study provides some evidential support for the differential development of MFI in participants with varying levels of functional recovery following whiplash. Similar findings are noted by Karlsson et al. [55] and Abbott et al. [56], when comparing 31 individuals with severe disability (chronic WAD lasting >6 months and <3 years) to healthy controls.
In a cross-sectional study by Pedler et al. [57] comparing 43 individuals with chronic WAD (>3 months and <10 years) and 16 healthy controls, the authors found no significant differences in MFI in the soleus muscle between people with chronic WAD and a demographically similar asymptomatic control group, despite between-group differences in MFI at the cervical multifidus – suggesting the potential of local mechanisms at the cervical spine contributing to the differences noted.
The mechanism behind how these aforementioned findings might contribute to the clinical presentation of WAD remains to be elucidated. Nevertheless, it is conceivable that disuse; as a consequence of fear-avoidance behaviour [58] and passive coping strategies [59], which has been shown to produce reductions in muscle volume as well as intramuscular fatty infiltration [60, 61], may play a role in the development of the notable changes in muscle morphology.
Central sensitisation
Where peripheral sensitisation is often associated with acute WAD, increasing evidence exists to suggest that prolonged noxious input manifests as central sensitisation; the amplification of neural signaling within the central nervous system that results in pain hypersensitivity, and that these changes can remain long after nociceptive input has disappeared. [62, 63]
The two hallmark characteristics of central sensitisation are the presence of allodynia (pain due to a stimulus that does not usually provoke pain) and hyperalgesia (increased pain from a stimulus that usually provokes pain). While the mechanism of central sensitisation is still not well understood, the processes involved in this phenomenon result in increased responsiveness to a variety of stimuli includingmechanical pressure [64],
chemical substances [65],
cold temperature [66],
heat temperature [67], and
electrical stimuli. [64, 68]When the central nervous system is sensitised, tissue damage is not required to induce pain. This may explain the discrepancy between the absence of tissue damage and persistent pain complaints in chronic WAD patients. [69]
It is unclear when the central nervous system starts sensitising and when general widespread hypersensitivity appears. Four studies suggest that central sensitisation occurs three to six months after the initial onset of WAD. [66, 70–72 ] At the same time, it is important to recognize that the chronic WAD population is heterogeneous and that central sensitisation is not present in all WAD patients. [73]
As it relates to alterations in structural brain morphology, one study examining structural abnormalities shortly after MVC exposure (within two days), found no signs of edema or lesion in the acute WAD group when compared with healthy controls, nor could a prediction of symptom development be made. [74] Obermann et al. [75] conducted a cross-sectional case-control study that performed voxelbased morphometry in WAD patients with post-traumatic headache and neck pain within 14 days of MVC exposure. The authors found no structural brain alterations in the acute phase (initial 14 days). However, in those patients that developed chronic headache lasting longer than three months, decreased grey matter volume in the anterior cingulate cortex (ACC) and dorsolateral prefrontal cortex (DLPFC) was observed, and resolved after one year – coinciding with the cessation of headache symptoms. Another study analyzing the ventricle-brain ratio (VBR), found no difference in VBR between patients with chronic WAD and healthy controls. [76]
As it relates to alterations in brain function, a cross-sectional case-control study conducted by Freitag et al. [77] examining 17 subjects; five symptomatic chronic WAD patients (duration 14 to 34 months), five asymptomatic WAD patients, and seven healthy volunteers, all with no evidence of structural brain damage, confirmed by T2-weighted magnetic resonance imaging (MRI), found that chronic WAD patients showed a significantly decreased performance in psychophysical tasks of coherent motion detection and corresponding functional MRI (fMRI) activation in the middle temporal (MT) and middle superior temporal (MST) regions of the brain, which are known to be important cortical sites of visual motion processing, compared to asymptomatic patients and healthy volunteers. Linnman et al. [78] aimed to explore whether Neurokinin 1–receptor (NK1R) availability is altered in chronic pain patients as compared to healthy controls, and whether changes in NK1R expression are related to behavioural aspects of chronic pain.
The NK1R is a member of the tachykinin receptor family that preferentially binds to Substance P (SP); the neuropeptide that regulates affective behaviour, emesis, and nociception [79], and both NK1 and SP have been implicated in locomotive activity [80] and in pain processing. [81]
The authors found a decrease of NK1R availability in chronic WAD patients, and observed a negative correlation between kinesiophobia and NK1R availability per patient scores on the self-reported Tampa Scale for Kinesiophobia (TSK) Questionnaire. That is to say, patients found to have decreased NK1R density also had higher TSK scores; reflecting increased pain-related fear and avoidance behaviour.
Five studies examined alterations in brain perfusion/ metabolism through single-photon emission tomography (SPECT) and positron emission tomography (PET) imaging [82–86], of which, only Radanov et al. [82] found no indication for changes of brain perfusion. Sundström et al. [83] demonstrated that patients with chronic idiopathic neck pain showed decreased regional cerebral blood flow (rCBF) compared to healthy controls, which was most obvious in the parahippocampal and temporal regions, and the cerebellum. However, no such alterations could be observed in patients suffering from chronic WAD compared with healthy controls. Linnman et al. [84] found alterations in the left and right parahippocampal gyrus, left lingual gyrus, left and right posterior cingulate gyrus, right caudate nucleus and right pulvinar nucleus of the thalamus in chronic WAD patients. Bakhtadze et al. [85] found decreased perfusion of the parietal and frontal regions in patients with moderate to severe chronic idiopathic neck pain symptoms when compared to patients with only mild symptoms. Lastly, Lorberboym et al. [86] found regional CNS perfusion abnormalities by SPECT in 13/20 chronic WAD patients (range six months to five years).
However, these abnormalities were not equal for all patients. In 8 patients, perfusion abnormalities were observed in the temporal lobes, in three patients in the occipital lobes, in two patients in the frontal lobes, and another two patients in the basal ganglia.
Therefore, while some evidence exists demonstrating alterations in brain structure, function, perfusion and metabolism in individuals with chronic neck pain, the nature and location of these alterations is not entirely clear. Contradictory findings also exist, suggesting that multiple mechanisms may be responsible for the brain’s neuroplasticity associated with the perception of pain.
The patient’s psychological state
WAD patients may experience considerable psychological strain. In a meta-analysis of 24 studies, involving 4,502 patients, elevated psychological distress was associated with WAD with a large effect size (Cohen’s d = 0.90); appreciably larger than that observed amongst patients with spinal cord injuries (d = 0.69) or mild to moderate traumatic brain injury (d = 0.23). [87] Moreover, evidence exists suggesting that patients with persistent WAD are more likely to have had a pre-MVC history of psychiatric morbidity. [88]
Pain catastrophising; a person’s “tendency to magnify the threat value of pain stimulus and to feel helpless in the context of pain, and by a relative inability to inhibit pain-related thoughts in anticipation of, during or following a painful encounter,” [89] has been linked with poorer clinical outcomes in WAD patients. [90–95]
Extensive research has also been published on the impact of patient expectations with respect to recovery from WAD. [96–106] Throughout these studies, the expectations most commonly studied were expectation of pain resolution and expectation of return to work; and in each study, patients’ initial expectations of recovery were outcome-predictive. [96–100, 103, 106] In one cohort study involving 6,015 adults with WAD, those who expected to recover, recovered at more than three times the pace of those who did not anticipate recovery. [99] In another study, expectation of recovery was found to predict resumption of not just work, but also routine engagement in activities. [104] Expectation of recovery has also been shown to serve as a mediator of pain catastrophising and fear of movement. [97] Consequently, possessing higher levels of self-efficacy has been shown to be associated with enhanced physical functioning and health status, and lower pain intensity, perceived disability, depressive symptoms and fatigue in individuals with chronic musculoskeletal pain. [107]
The impact of compensation
The legal concept of pain and suffering emerged from the origins of civil tort law in early England, and Western society has long recognized that compensation for a loss caused by another is an acceptable effort to make one whole. [108] Yet, it is hypothesised that observing WAD as a compensable phenomenon may result in worse health outcomes. [109] This understanding stems from the belief that fault-based compensation systems are harmful to health because such systems require individuals to prove poor health and functional decline in an adversarial environment, which, in turn, is thought to negatively affect recovery. [110] Moreover, the prospect of financial gain is presumed to motivate some individuals to exaggerate the severity of their symptoms or health status. [111]
These views were corroborated by a population-based study of 7,462 road traffic accident claimants, conducted by Cassidy et al. [112], that saw a 28% reduction in the incidence of whiplash claims along with a reduction in the median time to close claims by more than 200 days, despite increases in the number of vehicle-damage claims and the number of kilometers driven after the province of Saskatchewan switched to a no-fault insurance system from its previous fault-based system. Similar findings were reported in the state of Victoria, Australia, following the introduction of legislation limiting court actions and compensation for whiplash. [113] In keeping with this theme, Cameron et al. [114] conducted an interrupted time series to assess whether a change in legislation improved health status and quality of life for people with whiplash in New South Wales, Australia. The authors compared three independent groups at baseline (after injury and lodgement of the insurance claim) and two years later. The first group included whiplash claimants who were involved in a motor vehicle crash in the period July to September 1999, before the legislative change. The second group included whiplash claimants who were exposed to MVC during the period July to December 2001, approximately two years after the legislative change. The third group included whiplash claimants who were exposed to MVC between July 2003 and March 2004. Adjusted analysis for the three groups to control for additional factors such as age or gender, and other possible confounders revealed recovery rates of 37%, 52% and 49% in groups 1, 2, and 3, respectively. Reductions in pain intensity were also significantly improved (p = 0.03) in groups 2 and 3 compared to group 1, as were improvements in physical component scores (p = 0.001) and health status (p = 0.01), measured by SF-36 – demonstrating that health outcomes for people with whiplash were substantially improved after legislative change that restricted access to compensation for noneconomic loss, introduced clinical guidelines for the management of whiplash and provided earlier acceptance of compensation claims and greater provision of early treatment.
The notion that regional sociocultural factors may influence patterns of behaviour led Schrader and colleagues to study the natural evolution of WAD in Lithuania; a country where, at the time, most drivers did not have personal injury insurance, and with little public awareness, even among Lithuanian doctors, about whiplash and its potential to cause disability. [115] The authors conducted a retrospective questionnaire-based cohort study of 202 individuals and found that the incidence of chronic neck pain was no higher among individuals involved in MVCs than those in the general population. While recall bias may account for the study findings, the data noted comports with the evidence that individual expectancies; which are not only influenced by one’s hardiness, but also through social learning, [116] duly predict outcomes. In a subsequent cohort study of 210 Lithuanian motorists involved in MVCs conducted by Obelieniene and colleagues, the authors found no significant differences between motorists involved in rear end collisions and the control group concerning frequency and intensity of neck pain, and headache symptoms after one year. [117]
Rydman et al. [118] offer further support for the compensation hypothesis based on a longitudinal cohort study of 144 individuals reporting neck pain after being involved in a motor vehicle collision. The authors noted a higher non-recovery rate among individuals who filed insurance claims (73%) compared to those who did not (51%) over a period of two to four years. The authors further noted an overrepresentation of patients with elevated levels of mental distress at the time of the collision that may have influenced their compensation-seeking behaviour.
Other factors
In addition to the aforementioned negative risk factors associated with poor WAD prognosis, namely; catastrophisation, and compensation and legal factors, some evidence exists to suggest that post-MVC pain and disability [119–125], and early use of healthcare [121] negatively affect WAD prognosis.
Interestingly, factors identified as not being associated with WAD prognosis include post-MVC MRI or radiologic findings [121, 126], motor dysfunctions [127], and collision factors; such as the direction of impact, use of seatbelts or headrests, and the speed of the vehicle at the time of impact. [119, 121, 122, 124] The clinical presentation and prognosis of WAD are also not affected by pre-existing disc degeneration. [21] This notion is further supported by a prospective ten-year follow-up study that compared WAD patients and asymptomatic subjects using MRI. The study revealed that progressive decreases in the signal intensity of the intervertebral discs was observed more frequently among WAD patients than the healthy controls, while structural changes in the cervical spine, including posterior disc protrusion, disc space narrowing, and foraminal stenosis, progressed almost equally in both groups. Furthermore, the clinical symptoms observed in both the WAD patients and the healthy controls were not associated with any of the MRI findings. These findings suggest that, although some WAD patients may suffer from long-lasting clinical symptoms, whiplash exposure may not necessarily accelerate the symptomatic structural deterioration of the cervical spine. Instead, the progression of disc degeneration observed in the majority of WAD patients might be attributed to the normal ageing process, similar to the changes seen in the healthy controls. [128] Another prospective 11–year follow-up study that compared the incidence and prevalence of Modic changes in the cervical spines of WAD patients compared with healthy controls, found that Modic changes were not related to clinical symptoms. Instead, the development of new Modic changes was significantly associated with age, heavy labour, and preexisting disc degeneration, but not with the details of the MVCs. [129]
Discussion
Establishing a causal inference between a trauma mechanism and WAD
Where a disease denotes a condition characterised by functional impairment, structural change, and the presence of specific signs and symptoms, a disorder is defined by functional impairment without structural change. [130] Consequently, a diagnosis is the informed opinion of a clinician who provides a label to the patient advising them of their condition. A widely accepted definition of causation is that a specific occurrence serves as an antecedent event or condition that was necessary for the development of a specific condition or injury at the moment that it occurred, given that other circumstances are fixed. [131] In order to ascertain the nosological classification of WAD, a causal relationship linking MVC exposure to symptom development that comports with detectable evidence of one or more of the aforementioned suggested mechanisms of WAD development must be constituted. This is achieved through consideration of the nine principles, proposed by Bradford Hill, in determining whether a causal inference is reasonable. [16] The practical application of Hill’s criteria is especially useful in determining the credibility of a cause-and-effect relationship in a forensic setting (in individuals evaluated for a legal matter) in order to answer two major forensic questions: “could the exposure have caused the injury or condition outcome in this case?” and “did the exposure cause the injury or condition outcome in this case?”
Herein, the application and interpretation of each of Hill’s criteria, as they pertain to the etiology and pathology of WAD, is considered.Strength of association refers to a strong statistical association for causality. Nolet et al. conducted a systematic review and meta-analysis to examine the association between MVC-related neck injury and future neck pain compared to the prevalence of neck pain in the general population, and calculated an unadjusted relative risk of future neck pain in the MVC-exposed population with neck injury of 2.3; reflecting a 57% attributable risk. [132] At face value, this finding demonstrates a positive association between MVC-related neck injury and future neck pain. However, the broad operational definition of injury applied by the authors: “self-reported injury, primary care or emergency room physician diagnosed injury or an injury that had been filed with an automobile insurance company” [132] raises the risk of overestimating injury incidence. If we were to apply the injury definition provided in this paper, a strong statistical association between MVC exposure and WAD would be less likely.
Consistency means that several studies involving various groups of patients produce the same conclusion. WAD has been described among distinct populations with significant differences in its incidence and prevalence. [112–116, 133, 134] There is also considerable variation in the clinical presentation of WAD; owing to a lack of consistency.
Specificity refers to the degree to which an exposure derives a particular effect. For example; the most common mechanism of lateral ankle sprain is excessive inversion and internal rotation of the hindfoot while the leg is in external rotation, placing maximal strain on the lateral ankle ligaments. [135] Conversely, the descriptive validity of WAD; the degree to which it can be distinguished from other similar conditions as a result of a specific cause, is inadequate. [136, 137]
Temporality, described as the obvious principle that cause must precede effect, is perhaps the sole universally agreed upon principle deemed essential for causal inference – where symptom development weeks after a collision would refute causation. At the same time, temporal subsequence does not necessarily imply consequence; as illustrated in an elaborately-designed study by Castro et al., where 51 healthy adults underwent a mock collision that resulted in almost 20% of the subjects indicating “whiplash-like” symptoms within three days of exposure. [138] None of the test subjects raised any doubts about having been in a real collision after being exposed to 0.03 g-force; equivalent to four to five times less than the force applied when a person takes their first step to initiate walking. Yet, symptoms were reported and attributed to this exposure, which lacked the biomechanical potential to induce injury. In this example, where the timing of symptom development may adhere to the principle of temporality, the development of symptoms in the absence of trauma violates the principles of strength of association, consistency, specificity, biological gradient, biological plausibility, coherence, experiment and analogy.
Biological gradient (dose-response relationship) – Hill wrote that “if a dose response is seen, it is more likely that the association is causal.” [16] Meaning that greater exposure should generally lead to an increased incidence of the effect. Some authors suggest that a “limit of harmlessness” in low-velocity collisions (<15km/h) lies between 10–15km/h, [139] whereas Davis argues that although vehicle damage may not occur up until impacts reach speeds of 15km/h, 4km/h impacts can induce physical injury. [140] Giannoudis et al. conducted a retrospective study of 101 consecutive polytrauma patients at a Level I Trauma Centre, and found that the incidence of whiplash is relatively low (13%) following high-energy trauma; concluding that “there is no dose-response relationship between the magnitude of trauma severity and incidence of whiplash injury.” [141] In this study, whiplash injury was defined according to the Quebec Task Force guidelines as neck pain, stiffness, or headache following the original inciting event, and trauma severity was defined in the following ways: high-energy trauma; reflecting a fall from a height of more than two metres, high-velocity road traffic accidents; reflecting collisions occurring at speeds greater than 30km/h, and an Injury Severity Score (ISS) >16; reflecting major trauma. [142, 143]
Biological plausibility means that the association between cause and effect must appear reasonable based on our current biological knowledge. The primary concern regarding WAD pathogenesis is the lack of detectable tissue pathology to substantiate the notion that a traumatic mechanism accounts for the development of post-MVC symptoms. This understanding by no means discounts the patient’s pain experience, but rather highlights the impact that psychosocial factors and expectancies associated with WAD can have on symptom chronicity and the patient’s perception of disability and recovery.
Coherence is viewed as being similar to biological plausibility where “the cause-and-effect interpretation of our data should not seriously conflict with the generally known facts of the natural history and biology of the disease.” [16] The principle of coherence is not satisfied in the absence of gross, biochemical or histopathological evidence to substantiate a mechanistic construct for the development of WAD.
Experiment – Hill explained that evidence drawn from experimental manipulation may lead to the strongest support for causal inference. Implementation of this strategy has been attempted by way of several in vitro biomechanical, and in vivo animal studies demonstrating collagen fibre disorganisation and/or disruption and hyper-laxity [25–28], in addition to axonal swelling changes, hyper-excitability, and permanent alterations in neuronal signaling within the spinal cord. [29–32] However, the effects noted are generally imperceptible in live humans, negating this principle’s utility in drawing a cause-and-effect relationship.
Analogy has been interpreted to mean that when one causal agent is known, the standards of evidence are lowered for a second causal agent that is similar in some way. [144] The cause-and-effect relationship between a trauma mechanism and the development of WAD is generally rejected, as this phenomenon violates the common traumatological and wound healing principles that inform our current understanding of hard and soft tissue injuries. [145–147] In application; if we once again consider lateral ankle sprain – the primary tissue injury (anterior talofibular ligament and/or sometimes the calcaneofibular ligament) is easily detectable, the mechanism of injury is well-defined, there is a clear pathomechanical impairment associated with the condition, and a significant improvement in mechanical stability is realised for the majority of individuals within a timeline that comports with the normative processes of wound healing. [148] If we attempt to infer a trauma mechanism for the development of WAD, and predict its prognosis using this principle of analogy, we fall short given our inability to 1) detect structural tissue damage and 2) correlate the persistence of symptoms with a narrative that supports the notion that physical injury accounts for the patient’s condition. This line of reasoning is further corroborated by the findings noted in a retrospective survey investigating the occurrence of acute and chronic neck pain in demolition derby drivers. Of the 40 derby drivers surveyed, reporting exposure to a median of 1,632 lifetime collisions, with mean and maximum collision speeds of 41.6 km/h and 72 km/h, respectively, only two reported their worst post-participation neck pain lasting more than three months, and for one it lasted more than a year. For the majority, the worst neck pain event lasted less than 21 days. [149] These findings suggest that motivational differences between derby drivers and people sustaining whiplash injuries in the general population may account for outcome differences. While this study is at risk of both recall and selection bias, as a group, the derbyists demonstrated that they were less likely to succumb to perceptions of victimhood or illness behaviour and symptom amplification – characteristics known to prolong recovery. Habituation; a form of nonassociative learning that refers to any decrease in innate responsiveness to a repeated stimulus, may also play a role in the derbyists responses to collision exposures. [150, 151]To this end, establishing a distinct cause-and-effect relationship between a trauma mechanism and the development of WAD via the aforementioned framework is untenable. As WAD is a disorder that is diagnosed clinically following a patient’s report of symptoms that are commonly attributed to the temporality of MVC exposure in the absence of detectable tissue pathology, it may then be viewed, primarily, as a social construct that is significantly impacted by psychosocial factors, which may amplify otherwise benign bodily symptoms, or transform the possibility of a minor injury into one that is viewed as serious – in turn generating anxiety that sets in motion the phenomenon of symptom expectation and self-imposed functional disability. Those who may reject this inference might employ the ad ignorantiam argument by retorting with the popular aphorism: “absence of evidence is not evidence of absence”. While there is some truth to this statement, absence of evidence may constitute evidence of absence when negative evidence has probative value to support a particular hypothesis over another, to the extent that negative evidence is more likely to draw a scientifically sound conclusion under the favoured hypothesis than under the alternative hypothesis. [152]
Limitations
Although a comprehensive summary regarding the evidence underlying the most commonly theorised biological and psychosocial mechanisms of WAD pathogenesis was attempted, a primary limitation of this review includes finite access to data, hence the use of a single search database. The author is also cognisant that employing a narrow string of search terms may have unintentionally missed other published research studies pertaining to some aspect of WAD pathology.
Conclusion
A definitive etiopathological pathway displaying a causal relationship between MVC exposure and WAD development remains to be elucidated. While the face validity of WAD is good; as both clinicians and patients recognise the condition, the evidence supporting the various purported constructs suggesting a causal link between a trauma mechanism and the development of symptoms is inadequate. In the absence of a defined injury mechanism, a sophisticated understanding of the interconnected nature of biological, psychological, and social states and processes involved in the perception of pain is recommended. Therefore, future research is required to develop a better understanding of how to enhance individuals’ expectations and abilities to adapt and self-manage in the face of physical, emotional, and social challenges, as this appears to significantly impact recovery.
Footnotes
The author has no disclaimers, competing interests, or sources of support or funding to report in the preparation of this manuscript.
REFERENCES
Holm, LW, Carroll, LJ, Cassidy, JD et al.
The Burden and Determinants of Neck Pain in Whiplash-associated Disorders
after Traffic Collisions: Results of the Bone and Joint Decade 2000–2010
Task Force on Neck Pain and Its Associated Disorders
Spine (Phila Pa 1976). 2008 (Feb 15); 33 (4 Suppl): S52-59Evans RW.
Persistent post-traumatic headache, postconcussion syndrome, and whiplash injuries:
the evidence for a non-traumatic basis with an historical review.
Headache. 2010;50(4):716–724.Crowe H.
A new diagnostic sign in neck injuries.
Calif Med. 1964;100:12–13.Spitzer WO, Skovron ML, Salmi LR, Cassidy JD, Duranceau J, Suissa S, Zeiss E.
Scientific Monograph of the Quebec Task Force on Whiplash-Associated Disorders
Redefining Whiplash and its Management
Spine (Phila Pa 1976). 1995 (Apr 15); 20 (8 Suppl): S1-S73Hincapie CA, Cassidy JD, Côté P, Carroll LJ, Guzman J.
Whiplash injury is more than neck pain: a population-based
study of pain localization after traffic injury.
J Occup Environ Med. 2010;52(4):434–440.Van Goethem JW, Biltjes IG, van den Hauwe L, Parizel PM, De Schepper AM.
Whiplash injuries: Is there a role for imaging?
Eur J Radiol. 1996;22(1):30–37.Panjabi MM, Cholewicki J, Nibu K, Grauer J, Vahldiek M.
Capsular ligament stretches during in vitro whiplash simulations.
J Spinal Disord. 1998;11:227–232.Winkelstein BA, Nightingale RW, Richardson WJ, Myers BS.
The cervical facet capsule and its role in whiplash injury:
a biomechanical investigation.
Spine. 2000;25:1238–1246.Panjabi MM, Ito S, Pearson AM, Ivancic PC.
Injury mechanisms of the cervical intervertebral disc during simulated whiplash.
Spine. 2004;29(11):1217–1225.Ivancic P, Pearson AM, Panjabi MM, Ito S.
Injury of the anterior longitudinal ligament during whiplash simulation.
Eur Spine J. 2004;13(1):61–68.Hernández IA, Fyfe KR, Heo G, Major PW.
The role of sternocleidomastoid muscle in simulated
low velocity rear-end impacts.
Euro Spine J. 2006;15(6):876–885.Sterling M.
Differential development of sensory hypersensitivity and a measure
of spinal cord hyperexcitability following whiplash injury.
Pain. 2010;150(3):501–506.Buitenhuis J, de Jong PJ, Jaspers JP, Groothoff JW.
Catastrophizing and causal beliefs in whiplash.
Spine. 2008;33(22):2427–2433.Richter M, Ferrari R, Otte D, Kuensebeck HW, Blauth M, Krettek C.
Correlation of clinical findings, collision parameters, and psychological
factors in the outcome of whiplash associated disorders.
J Neurol Neurosurg Psychiatry. 2004;75:758–764.Stovner L.
The nosologic status of the whiplash syndrome:
a critic review based on a methodological approach.
Spine. 1996;21(23):2735–2746.Hill AB.
The environment and disease: association and causation.
Proc R Soc Med. 1965;58:295–300.Höfler M.
The Bradford Hill considerations on causality:
a counterfactual perspective.
Emerg Themes Epidemiol. 2005;2:11.Schünemann H, Hill S, Guyatt G, Akl EA, Ahmed F.
The GRADE approach and Bradford Hill’s criteria for causation.
J Epidemiol Commun Health. 2011;65:392395.Howick J, Glasziou P, Aronson JK.
The evolution of evidence hierarchies:
what can Bradford Hill’s ‘guidelines for causation’ contribute?
JRSM. 2009;102(5):186–194.Daimon K, Fujiwaa H, Nishiwaki Y, Okada E, Nojiri K, Shimizu K, Ishihama H.
A 20-year prospective longitudinal MRI study on cervical spine
after whiplash injury: Follow-up of a cross-sectional study.
J Orthop Sci. 2019;24(4):579–583.Chung NS, Jeon CH, Lee YS, Park JH, Lee HD.
Is preexisting cervical disk degeneration a prognostic
factor in whiplash-associated disorders?
Clin Spine Surg. 2017;30(9):1251–1255.Ichihara D, Okada E, Chiba K, Toyama Y, Fujiwara H, Momoshima S, Nishiwaki Y.
Longitudinal magnetic resonance imaging study on
whiplash injury patients: minimum 10-year follow-up.
J Orthop Sci. 2009;14(5):602–610.Vetti N, Krĺkenes J, Eide GE, Rřrvik J, Gilhus NE, Espeland A.
Are MRI high-signal changes of alar and transverse ligaments
in acute whiplash injury related to outcome?
BMC Musculoskelet Disord. 2010;11(11):260–267.Vetti N, Krĺkenes J, Eide GE, Rřrvik J, Gilhus NE, Espeland A.
MRI of the alar and transverse ligaments in whiplash-associated disorders
(WAD) grades 1–2: high-signal changes by age, gender, event and time since trauma.
Neuroradiology. 2009;51(4):227–2235.Ivancic PC, Ito S, Tominaga Y, Rubin W, Coe MP, Ndu AB, Carlson EJ, Panjabi MM.
Whiplash causes increased laxity of cervical capsular ligament.
Clin Biomech. 2008;23(2):159–165.Steilen D, Hauser R, Woldin B, Sawyer S.
Chronic neck pain: making the connection between
capsular ligament laxity and cervical instability.
Open Orthop J. 2014;8:326–345.Quinn KP, Bauman JA, Crosby ND, Winkelstein BA.
Anomalous fiber realignment during tensile loading of the rat facet capsular
ligament identifies mechanically induced damage and physiological dysfunction.
J Biomech. 2010;43(10):1870–1875.Quinn KP, Lee KE, Ahaghotu CC, Winkelstein BA.
Structural changes in the cervical facet capsular ligament:
potential contributions to pain following subfailure loading.
Stapp Car Crash J. 2007;51:169–187.Dong L, Odeleye AO, Jordan-Sciutto KL, Winkelstein BA.
Painful facet joint injury induces neuronal stress activation in the DRG:
implications for cellular mechanisms of pain.
Neurosci Lett. 2008;443(2):90–94.Kallakuri S, Singh A, Lu Y, Chen C, Patwardhan A, Cavanaugh JM.
Tensile stretching of cervical facet joint capsule
and related axonal changes.
Eur Spine J. 2008;17(4):556–563.Lu Y, Chen C, Kallakuri S, Patwardhan A, Cavanaugh JM.
Neural response of cervical facet joint capsule to stretch:
a study of whiplash pain mechanism.
Stapp Car Crash J. 2005;49:49–65.Quinn KP, Dong L, Golder FJ, Winkelstein BA.
Neuronal hyperexcitability in the dorsal horn after painful facet joint injury.
Pain. 2010;151(2):414–421.Lord SM, Barnsley L, Wallis BJ, Bogduk N.
Third occipital nerve headache: a prevalence study.
J Neurol Neurosurg Psychiatry. 1994;57(10):1187–1190.Barnsley L, Lord SM, Wallis BJ, Bogduk N.
The Prevalence of Chronic Cervical Zygapophysial Joint Pain After Whiplash
Spine (Phila Pa 1976) 1995 (Jan 1); 20 (1): 20–26Lord SM, Barnsley L, Wallis BJ, Bogduk N.
Chronic Cervical Zygapophysial Joint Pain After Whiplash:
A Placebo–Controlled Prevalence Study
Spine (Phila Pa 1976) 1996 (Aug 1); 21 (15): 1737–1744Lord SM, Barnsley L, Wallis BJ, McDonald GJ, Bogduk N.
Percutaneous radio-frequency neurotomy for chronic
cervical zygapophyseal-joint pain.
N Engl J Med. 1996;335(23):1721–1726.Lord SM, Barnsley L, Bogduk N.
Percutaneous radiofrequency neurotomy in the treatment
of cervical zygapophysial joint pain: a caution.
Neurosurg. 1995;36:732–739.McDonald GJ, Lord SM, Bogduk N.
Long-term followup of patients treated with cervical
radiofrequency neurotomy for chronic neck pain.
Neurosurg. 1999;45(1):61–68.Govind J, King W, Bailey B, Bogduk N.
Radiofrequency neurotomy for the treatment of third occipital headache.
J Neurol Neurosurg Psychiatry. 2003;74(1):88–93.Barnsley L.
Percutaneous radiofrequency neurotomy for chronic neck pain:
outcomes in a series of consecutive patients.
Pain Med. 2005;6(4):282–286.Smith AD, Jull G, Schneider G, Frizzell B, Hooper RA, Dunne-Proctor R, Sterling M.
Cervical radiofrequency neurotomy reduces psychological
features in individuals with chronic whiplash symptoms.
Pain Phys. 2014;17(3):265–274.Boswell MV, Manchikanti L, Kaye AD, Bakshi S, Gharibo CG, Gupta S.
A best-evidence systematic appraisal of the diagnostic accuracy and
utility of facet (zygapophysial) joint injections in chronic spinal pain.
Pain Phys. 2015;18:E497–E533.Gellhorn AC, Katz JN, Suri P.
Osteoarthritis of the spine: the facet joints.
Nat Rev Rheumatol. 2012;9(4):216–224.Kim JH, Sharon A, Cho W, Emam M, Hagen M, Kim SY.
The prevalence of asymptomatic cervical and lumbar facet arthropathy:
a computed tomography study.
Asian Spine J. 2019;13(3):417–422.Manchikanti L, Manchikanti KN, Cash KA, Singh V, Giordano J.
Age-related prevalence of facet-joint involvement in chronic neck and low back pain.
Pain Phys. 2008;11:67–75.Huygen F, Kallewaard JW, Tulder M, Van Boxem K, Vissers K, Kleef M.
“Evidence-Based Interventional Pain Medicine According to
Clinical Diagnoses”: update 2018.
Pain Practice” 2019;19(6):664675.Fenton BW, Shih E, Zolton J.
The neurobiology of pain perception in normal and persistent pain.
Pain Manag. 2015;5(4):297–317.Robertson LS.
Injury epidemiology. 3rd ed.
New York: Oxford University Press; 2007. p. 9.Lumley MA, Cohen JL, Borszcz GS, Cano A, Radcliffe AM, Porter LS.
Pain and emotion: a biopsychosocial review of recent research.
J Clin Psychol. 2011;67(9):942–968.Scott S, Sanderson PL.
Whiplash: a biochemical study of muscle injury.
Eur Spine J. 2002;11(4):389–92.Peolsson A, Peterson G, Trygg J, Nilsson D.
Multivariate analysis of ultrasound-recorded dorsal strain sequences:
Investigation of dynamic neck extensions in women with chronic whiplash associated disorders.
Sci Rep. 2016;6:1–11.Peterson G, Nilsson D, Trygg J, Falla D, Dedering A, Wallman T, Peolsson A.
Novel insights into the interplay between ventral neck muscles
in individuals with whiplash-associated disorders.
Sci Rep. 2015;5:1–13.Farron J, Varghese T, Thelen DG.
Measurement of tendon strain during muscle twitch contractions using ultrasound elastography.
IEEE Trans Ultrason Ferroelectr Freq Control. 2009;56(1):27–35.Elliott JM, Courtney DM, Rademaker A, Pinto D, Sterling MM, Parish TB.
The rapid and progressive degeneration of the cervical multifidus in whiplash:
an MRI study of fatty infiltration.
Spine. 2015;40(12):694–700.Karlsson A, Leinhard OD, Ĺslund U, West J, Romu T, Smedby Ö, Zsigmond P.
An investigation of fat infiltration of the multifidus muscle in patients
with severe neck symptoms associated with chronic whiplash-ssociated disorder.
J Orthop Sports Phys Ther. 2016;46(10):886–893.Abbott R, Peolsson A, West J, Elliott JM, Ĺslund U, Karlsson A, Leinhard OD.
The qualitative grading of muscle fat infiltration in whiplash
using fat and water magnetic resonance imaging.
Spine J. 2018;18(5):717725.Pedler A, McMahon K, Galloway G, Durbridge G, Sterling M.
Intramuscular fat is present in cervical multifidus but not
soleus in patients with chronic whiplash associated disorders.
PLoS One. 2018;13(5):1–10.Vlaeyen J, Linton S.
Fear-avoidance model of chronic musculoskeletal pain: 12 years on.
Pain. 2012;153(6):1144–1147.Carroll LJ, Ferrari R, Cassidy JD, Côté P.
Coping and recovery in whiplash-associated disorders: early use of passive
coping strategies is associated with slower recovery of neck pain and pain-related disability.
Clin J Pain. 2014;30(1):1–8.Manini TM, Clark BC, Nalls MA, Goodpaster BH, Ploutz-Snyder LL.
Reduced physical activity increases intermuscular adipose tissue in healthy young adults.
Am J Clin Nutr. 2007;85(2):377–384.Clark BC.
In Vivo Alterations in skeletal muscle form and function after disuse atrophy.
Med Sci Sports Exerc. 2009;41(10):1869–1875.Woolf CJ.
Evidence for a central component of postinjury pain hypersensitivity.
Nature. 1983;306:686–688.Woolf CJ.
Central sensitization: implications for the diagnosis and treatment of pain.
Pain. 2011;152:1–31.Desmeules JA, Cedraschi C, Rapiti E, Baumgartner E, Finckh A.
Neurophysiologic evidence for a central sensitization in
patients with fibromyalgia.
Arthritis Rheum. 2003;48(5):1420–1429.Morris VH, Cruwys SC, Kidd BL.
Characterisation of capsaicin-induced mechanical hyperalgesia as a marker
for altered nociceptive processing in patients with rheumatoid arthritis.
Pain. 1997;71:179–186.Kasch H, Qerama E, Bach FW, Jensen TS.
Reduced cold pressor pain tolerance in non-recovered whiplash patients:
a 1-year prospective study.
Eur J Pain. 2005;9(5):561–569.Meeus M, Nijs J, Van De Wauwer N, Toeback L, Truijen S.
Diffuse noxious inhibitory control is delayed in chronic
fatigue syndrome: an experimental study.
Pain. 2008;139:439–448.Banic B, Petersen-Felix S, Andersen OK, Radanov BP, Villiger PM, Arendt-Nielsen L.
Evidence for spinal cord hypersensitivity in chronic pain
after whiplash injury and in fibromyalgia.
Pain. 2004;107(1–2):7–15.Herren-Gerber R, Weiss S, Arendt-Nielsen L, Petersen-Felix S.
Modulation of central hypersensitivity by nociceptive input
in chronic pain after whiplash injury.
Pain Med. 2004;5:366–376.Sterling M, Jull G, Vicenzino B, Kenardy J.
Sensory hypersensitivity occurs soon after whiplash injury
and is associated with poor recovery.
Pain. 2003;104:509–517.Sterling M, Pedler A, Chan C, Puglisi M, Vuvan V, Vicenzino B.
Cervical lateral glide increases nociceptive flexion reflex threshold
but not pressure or thermal pain thresholds in chronic whiplash
associated disorders: as pilot randomised controlled trial.
Man Ther. 2010;15(2):149–153.Chien A, Eliav E, Sterling M.
The development of sensory hypoesthesia after whiplash injury.
Clin J Pain. 2010;26(8):722–728.Nijs J, Van Houdenhove B, Oostendorp RA.
Recognition of central sensitization in patients with musculoskeletal pain:
Application of pain neurophysiology in manual therapy practice.
Man Ther. 2010;15:135–141.Borchgrevink G, Smevik O, Haave I, Haraldseth O, Nordby A, Lereim I.
MRI of cerebrum and cervical columna within two days
after whiplash neck sprain injury.
Injury. 1997;28(5–6):331–335.Obermann M, Nebel K, Schumann C, Holle D, Gizewski ER, Maschke M.
Gray matter changes related to chronic posttraumatic headache.
Neurol. 2009;73(12):978–983.Sturzenegger M, Radanov BP, Winter P, Simko M, Farra AD, Di Stefano G.
MRI-based brain volumetry in chronic whiplash patients:
no evidence for traumatic brain injury.
Acta Neurol Scand. 2008;117(1):49–54.Freitag P, Greenlee MW, Wachter K, Ettlin TM, Radue EW.
fMRI response during visual motion stimulation in patients
with late whiplash syndrome.
Neurorehabil Neural Repair. 2001;15(1):31–37.Linnman C, Appel L, Furmark T, Söderlund A, Gordh T, Lĺngström B.
Ventromedial prefrontal neurokinin 1 receptor availability is reduced in chronic pain.
Pain. 2010;149(1):64–70.Mantyh PW.
Neurobiology of substance P and the NK1 receptor.
J Clin Psychiatry. 2001;63:6–10.Elliott PJ, Iversen SD.
Behavioural effects of tachykinins and related peptides.
Brain Res. 1986;381:68–76.Woolf CJ, Salter MW.
Neuronal plasticity: Increasing the gain in pain.
Science. 2000;288:1765–1768.Radanov BP, Bicik I, Dvorak J, Antinnes J, von Schulthess GK, Buck A.
Relation between neuropsychological and neuroimaging findings
in patients with late whiplash syndrome.
J Neurol Neurosurg Psychiatry. 1999;66(4):485–489.Sundström T, Guez M, Hildingsson C, Toolanen G, Nyberg L, Riklund K.
Altered cerebral blood flow in chronic neck pain patients
but not in whiplash patients: a 99mTc-HMPAO rCBF study.
Eur Spine J. 2006;15(8):1189–95.Linnman C, Appel L, Söderlund A, Frans O, Engler H, Furmark T, Gordh T.
Chronic whiplash symptoms are related to altered regional
cerebral blood flow in the resting state.
Eur J Pain. 2009;13(1):65–70.Bakhtadze MA, Vernon H, Karalkin AV, Pasha SP, Tomashevskiy IO, Soave D.
Cerebral perfusion in patients with chronic neck and
upper back pain: preliminary observations.
J Manipulative Physiol Ther. 2012;35(2):76–85.Lorberboym M, Gilad R, Gorin V, Sadeh M, Lampl Y.
Late whiplash syndrome: correlation of brain SPECT with
neuropsychological tests and P300 event-related potential.
J Trauma. 2002;52(3):521–526.Craig A, Tran Y, Guest R, Gopinath B, Jagnoor J, Bryant RA, Collie A, Kenardy J.
Psychological impact of injuries sustained in motor vehicle crashes:
systematic review and meta-analysis.
BMJ Open. 2016;6(9):1–13.Kivioja J, Sjalin M, Lindgren U.
Psychiatric morbidity in patients with chronic whiplash-associated disorder.
Spine. 2004;29(11):1235–1239.Quartana PJ, Campbell CM, Edwards RR.
Pain catastrophizing: a critical review.
Expert Rev Neurother. 2009;9(5):745–758.Casey PP, Feyer AM, Cameron ID.
Course of recovery for whiplash associated disorders in a compensation setting.
Injury. 2015;46(11):2118–2129.Chiarotto A, Fortunato S, Falla D.
Predictors of outcome following a short multimodal rehabilitation program
for patients with whiplash associated disorders.
Eur J Phys Rehabil Med. 2015;51(2):133–141.De Pauw R, Coppieters I, Palmans T, Danneels L, Meeus M, Cagnie B.
Motor impairment in patients with chronic neck pain: does the
traumatic event play a significant role? A case-control study.
Spine J. 2018;18(8):1406–1416.Falla D, Peolsson A, Peterson G, Ludvigsson ML, Soldini E.
Perceived pain extent is associated with disability, depression and
self-efficacy in individuals with whiplash-associated disorders.
Eur J Pain. 2016;20(9):1490–1501.Ritchie C, Sterling M.
Recovery pathways and prognosis after whiplash injury.
J Orthop Sports Phys Ther. 2016;46(10):851–861.Smith AD, Jull GA, Schneider GM, Frizzell B, Hooper RA, Sterling MM.
Low pain catastrophization and disability predict successful outcome
to radiofrequency neurotomy in individuals with chronic whiplash.
Pain Pract. 2016;16(3):311–319.Carriere JS, Thibault P, Adams H, Milioto M, Ditto B, Sullivan MJL.
Expectancies mediate the relationship between perceived injustice and
return to work following whiplash injury: a 1-year prospective study.
Eur J Pain. 2017;21(7):1234–1242.Carriere JS, Thibault P, Milioto M, Sullivan MJL.
Expectancies mediate the relations among pain catastrophizing,
fear of movement, and return to work outcomes after whiplash injury.
J Pain. 2015;16(12):1280–1287.Carriere JS, Thibault P, Sullivan MJ.
The mediating role of recovery expectancies on the relation between depression and return-to-work.
J Occup Rehabil. 2015;25(2):348–356.Carroll LJ, Holm LW, Ferrari R, Ozegovic D, Cassidy JD.
Recovery in whiplash-associated disorders: do you get what you expect?
J Rheumatol. 2009;36(5):1063–1070.Elphinston RA, Thibault P, Carriere JS, Raineville P, Sullivan MJL.
Cross-sectional and prospective correlates of recovery expectancies
in the rehabilitation of whiplash injury.
Clin J Pain. 2018;34(4):306–312.Ferrari R, Louw D.
Correlation between expectations of recovery and injury severity perception in whiplash-associated disorders.
J Zhejiang Univ Sci B. 2011;12(8):683–686.Ozegovic D, Carroll LJ, Cassidy JD.
What influences positive return to work expectation?
Examining associated factors in a population-based cohort of whiplash=associated disorders.
Spine. 2010;35(15):708–713.Ozegovic D, Carroll LJ, David Cassidy J.
Does expecting mean achieving? The association between expecting to return to work
and recovery in whiplash associated disorders: a population-based prospective cohort study.
Eur Spine J. 2009;18(6):893–899.Söderlund A, Löfgren M, Stĺlnacke BM.
Predictors before and after multimodal rehabilitation for pain acceptance and engagement
in activities at a 1-year follow-up for patients with whiplash-associated disorders
(WAD)-a study based on the Swedish Quality Registry for Pain Rehabilitation (SQRP)
Spine J. 2018;18(8):1475–1482.Bialosky JE, Bishop MD, Cleland JA.
Individual expectation: an overlooked, but pertinent, factor in
the treatment of individuals experiencing musculoskeletal pain.
Phys Ther. 2010;90(9):1345–1355.Bostick GP, Carroll LJ, Brown CA, Harley D, Gross DP.
Predictive capacity of pain beliefs and catastrophizing in whiplash associated disorder.
Injury. 2013;44(11):14651471.Martinez-Calderon J, Zamora-Campos C, Navarro-Ledesma S, Luque-Suarez A.
The role of selfefficacy on the prognosis of chronic musculoskeletal pain:
a systematic review.
J Pain. 2018;19(1):10–34.Kulich RJ, Kreis PG, Fishman SM, Prescott JC, Jr, Pelletier NJ.
Forensic issues in pain: review of current practice.
Pain Pract. 2001;1(2):119135.Cameron P, Gabbe B.
The effect of compensation claims on outcomes after injury.
Injury. 2009;40:905–906.O’Donnell C.
Motor accident and workers’ compensation insurance design
for high quality health outcomes and cost containment.
Disabil Rehabil. 2000;22:88–96.Ferrari R, Kwan O, Russell AS, Pearce JM, Schrader H.
The best approach to the problem of whiplash? One ticket to Lithuania, please.
Clin Exp Rheumatol. 1999;17:321–326.Cassidy JD, Carroll LJ, Côté P, Lemstra M, Berglund A, Nygren Ĺ.
Effect of eliminating compensation for pain and suffering
on the outcome of insurance claims for whiplash injury.
N Engl J Med. 2000;342(16):1179–1186.Ferrari R, Russell AS.
Epidemiology of whiplash: an international dilemma.
Ann Rheum Dis. 1999;58:1–5.Cameron ID, Rebbeck T, Sindhusake D, Rubin G, Feyer AM, Walsh J, Schofield WN.
Legislative change is associated with improved health status in people with whiplash.
Spine. 2008;33(3):250–254.Schrader H, Obelieniene D, Bovim G, Surkiene D, Mickeviciene D, Sand T.
Natural evolution of late whiplash syndrome outside the medicolegal context.
Lancet. 1996;347(9010):1207–1211.Carroll LJ.
Beliefs and Expectations for Recovery, Coping, and Depression in
Whiplash-Associated Disorders: Lessening the Transition to Chronicity
Spine (Phila Pa 1976) 2011 (Dec 1); 36 (25 Suppl): S250–S256Obelieniene D, Schrader H, Bovim G, Miseviciene I, Sand T.
Pain after whiplash: a prospective controlled inception cohort study.
J Neurol Neurosurg Psychiatry. 1999;66:279–283.Rydman E, Ponzer S, Brisson R, Ottosson, Pettersson-Jarnbert H.
Long-term follow-up of whiplash injuries reported to insurance companies:
a cohort study on patient-reported outcomes and impact of financial compensation.
Eur Spine J. 2018;27:1255–1261.Walton DM, Macdermid JC, Giorgianni AA, Mascarenhas JC, West SC, Zammit CA.
Risk factors for persistent problems following acute whiplash injury:
update of a systematic review and meta-analysis.
J Orthop Sports Phys Ther. 2013;43(2):31–43.Goldsmith R, Wright C, Bell SF, Rushton A.
Cold hyperalgesia as a prognostic factor in
whiplash associated disorders: a systematic review.
Man Ther. 2012;17(5):402–410.Carroll, LJ, Holm, LW, Hogg-Johnson, S et al.
Course and Prognostic Factors for Neck Pain in Whiplash-associated
Disorders (WAD): Results of the Bone and Joint Decade 2000-2010
Task Force on Neck Pain and Its Associated Disorders
Spine (Phila Pa 1976). 2008 (Feb 15); 33 (4 Suppl): S83–92Kamper SJ, Rebbeck TJ, Maher CG, McAuley JH, Sterling M.
Course and prognostic factors of whiplash:
a systematic review and meta-analysis.
Pain. 2008;138(3):617–629.Williams M, Williamson E, Gates S, Lamb S, Cooke M.
A systematic literature review of physical prognostic factors
for the development of late whiplash syndrome.
Spine. 2007;32(25):E764– E780.Scholten-Peeters GG, Verhagen AP, Bekkering GE, van der Windt DA.
Prognostic factors of whiplash-associated disorders:
a systematic review of prospective cohort studies.
Pain. 2003;104(1–2):303–322.Côté P, Cassidy JD, Carroll L, Frank JW, Bombardier C.
A systematic review of the prognosis of acute whiplash and
a new conceptual framework to synthesize the literature.
Spine. 2001;26(19):E445–E458.Li Q, Shen H, Li M.
Magnetic resonance imaging signal changes of alar and transverse ligaments not
correlated with whiplash-associated disorders: a meta-analysis of case-control studies.
Eur Spine J. 2013;22(1):14–20.Daenen L, Nijs J, Raadsen B, Roussel N, Cras P, Dankaerts W.
Cervical motor dysfunction and its predictive value for long-term recovery
in patients with acute whiplash-associated disorders: a systematic review.
J Rehabil Med. 2013;45(2):113–122.Matsumoto M, Okada E, Ichihara D, Chiba K, Toyama Y, Fujiwara H, Momoshima S.
Prospective ten-year follow-up study comparing patients with whiplash-associated
disorders and asymptomatic subjects using magnetic resonance imaging.
Spine. 2010;35(18):16841690.Matsumoto M, Ichihara D, Okada E, Toyama Y, Fujiwara H, Momoshima S.
Modic Changes of the cervical spine in patients with whiplash injury:
a prospective 11-year follow-up study.
Injury. 2013;44(6):819–24.Condition, Disease, Disorder. AMA STYLE Insider. 2011.
[Accessed January 28, 2020]. from:
https://amastyleinsider.com/2011/11/21/condition-disease-disorder/#comments.Rothman KJ, Greenland S.
Causation and causal inference in epidemiology.
Am J Public Health. 2005;95(Suppl 1):S144–S150.Nolet PS, Emary PC, Kristman VL, Murnaghan K, Zeegers MP, Freeman MD.
Exposure to a Motor Vehicle Collision and the Risk of Future Neck Pain:
A Systematic Review and Meta-analysis
PM R. 2019 (Nov); 11 (11): 1228–1239Ferrari R, Constantoyannis C, Papadakis N.
Laypersons’ expectation of the sequelae of whiplash injury:
a crosscultural comparative study between Canada and Greece.
Med Sci Monit. 2003;9:120–124.Ferrari R, Lang C.
A cross-cultural comparison between Canada and Germany
of symptom expectation for whiplash injury.
J Spinal Disord Tech. 2005;18:92–97.Ferran NA, Oliva F, Maffulli N.
Ankle instability.
Sports Med Arthrosc Rev. 2009;17(2):139–145.Verhagen AP, Lewis M, Schellingerhout JM, Heymans MW, Dziedzic K, de Vet HC, Koes BW.
Do whiplash patients differ from other patients with
non-specific neck pain regarding pain, function or prognosis?
Man Ther. 2011;16(5):456–462.Radanov BP, Mannion AF, Ballinari P.
Are symptoms of late whiplash specific? A comparison of SCL-90-R symptom profiles
of patients with late whiplash and patients with chronic pain due to other types of trauma.
J Rheumatol. 2011;38(6):1086–1094.Castro WHM, Meyer SJ, Becke MER, Nentwig CG, Hein MF, Ercan BI, Thomann S, Wessels U, Du Chesne AE.
No stress – no whiplash?
Int J Leg Med. 2001;114:316.Castro WH, Schilgen M, Meyer S, Weber M, Peuker C, Wörtler K.
Do “whiplash injuries” occur in low-speed rear impacts?
Eur Spine J. 1997;6:366–375.Davis CG.
Rear-end impacts: vehicle and occupant response.
J Manipulative Physiol Ther. 1998;21:629– 639.Giannoudis PV, Mehta SS, Tsiridis E.
Incidence and outcome of whiplash injury after multiple trauma.
Spine. 2007;32(7):776–781.Palmer C.
Major trauma and the injury severity score – where should we set the bar?
Annu Proc Assoc Adv Automot Med. 2007;51:13–29.Palmer CS, Gabbe BJ, Cameron PA.
Defining major trauma using the 2008 Abbreviated Injury Scale.
Injury. 2016;47(1):109–115.Fedak KM, Bernal A, Capshaw ZA, Gross S.
Applying the Bradford Hill criteria in the 21st century:
how data integration has changed causal inference in molecular epidemiology.
Emerg Themes Epidemiol. 2015;12(14):1–9.Bahney CS, Zondervan RL, Allison P, Theologis A, Ashley JW, Ahn J, Miclau T, Marcucio RS, Hankenson KD.
Cellular biology of fracture healing.
J Orthop Res. 2019;37(1):35–50.Järvinen TA, Järvinen TL, Kääriäinen M, Kalimo H, Järvinen M.
Muscle Injuries.
Am J Sports Med. 2005;33(5):745–764.Cottrell JA, Turner JC, Arinzeh TL, O’Connor JP.
The biology of bone and ligament healing.
Foot Ankle Clin. 2016;21(4):739–761.Hubbard TJ, Hicks-Little CA.
Ankle ligament healing after an acute ankle sprain:
an evidence-based approach.
J Athl Train. 2008;43(5):523–529.Simotas AC, Shen T.
Neck pain in demolition derby drivers.
Arch Phys Med Rehabil. 2005;86:693–696.Grissom N, Bhatnagar S.
Habituation to repeated stress: get used to it.
Neurobiol Learn Mem. 2009;92(2):215224.Siegmund GP, Sanderson DJ, Myers BS, Inglis JT.
Rapid neck muscle adaptation alters the head kinematics of aware and unaware subjects
undergoing multiple whiplash-like perturbations.
J Biomech. 2003;36:473– 482.Thompson WC, Scurich N.
When does absence of evidence constitute evidence of absence?
Forensic Sci Int. 2018;291:e18–e19.
Return to WHIPLASH
Since 8-21-2022
| Home Page | Visit Our Sponsors | Become a Sponsor |
Please read our DISCLAIMER |